ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2014; 11(8):803-809. doi:10.7150/ijms.9084 This issue Cite
Research Paper
Integration of the Hybrid Adenoretroviral Vector AdLTR-luc Involves Both MoMLV Elements Flanking the Transgene
Changyu Zheng ![]() , Bruce J. Baum
, Bruce J. Baum
Molecular Physiology and Therapeutics Branch, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892-1190, USA
Abstract
Vector delivery is still a bottleneck for gene therapy. To overcome some disadvantages of adenoviral and retroviral vectors, we developed a hybrid vector. This hybrid vector, AdLTR-luc, was created by adding two elements from Moloney murine leukemia virus (MoMLV) flanking the luciferase cDNA into an E1/E3-deleted, replication deficient serotype 5 adenovirus vector (Zheng et al., Nature Biotechnol, 2000), and demonstrated that the MoMLV element upstream of the luciferase cDNA was broken during the integration event. The purpose of the current study was to determine if the MoMLV element downstream of the luciferase cDNA was also broken when integration occurred. We used the same A5 cell clones (#10 and 11) from the earlier the paper along with restriction endonuclease digestions, plus Southern hybridization, and PCR. Southern hybridization indicated that the luciferase cDNA was intact in the cloned cells. Results from Xho I and Sal I digestions showed that integration occurred in cloned cells. Southern hybridizations after Nco I digestion suggested that there was a break in both MoMLV elements, upstream and downstream of the luciferase cDNA. After DNA digestion with Not I, hybridization analyses indicated that the MoMLV upstream element was broken during integration. Digestion of genomic DNA with either Xba I/Kpn I, Bam HI/Sac I, or Bam HI/Nco I demonstrated that the MoMLV downstream element was also broken during integration. A PCR assay was unable to amplify the junctional region between the downstream MoMLV element and the adenoviral E2B gene, consistent with a break in that element. Although AdLTR-luc integration is atypical (Zheng et al., Nature Biotechnol, 2000), the present results suggest that both MoMLV elements have important roles in this event.
Keywords: Hybrid vector, integration, LTR, adenoretrovirus, gene therapy
Introduction
It has long been recognized that adenoviral/retroviral vector chimeras can be created to achieve high-efficiency stable transduction (1,2). In 2000, we reported the development of a novel hybrid adenoretroviral vector (3), in which we inserted 2.7 kb of Moloney murine leukemia virus (MoMLV) sequence 5´ to the luciferase cDNA (including, in a 5´ to 3´ direction, part of the envelope gene [1.5 kb], the 5´LTR [0.57 kb], and the packaging sequence [0.63 kb]) and 1 kb of MoMLV sequence 3´ to the luciferase cDNA (including ~0.5 kb, of the envelope gene and the intact 3´LTR). This hybrid vector, termed AdLTR-luc, still retained the adenoviral characteristic of highly efficient transduction and obtained the ability to mediate integration without the generation of retrovirus. Most importantly, results from PCR, Southern hybridization, fluorescence in situ hybridization, and gene walking analyses all demonstrated that this hybrid vector also obtained the retroviral-like characteristic of genomic integration, albeit atypical. Normally, adenoviruses integrate into a cell's genome very infrequently (10-3 to 10-5), resulting in unstable gene expression (4-8). Because of its ability to efficiently transduce cells, and mediate genomic integration, this hybrid vector may be useful for gene therapy applications.
Classically, following MoMLV entry into a cell, the RNA genome is reverse transcribed into DNA and integration of this DNA copy into a host cell chromosome occurs as an essential step in viral replication (9-11). The efficient integration of MoMLV requires two viral elements (11-18): the viral integrase (IN) and AATG sites located at the termini of the viral LTRs. Both the 5´ and 3´LTRs are considered necessary for the integration.
AdLTR-luc integration occurs in the absence of IN, suggesting that the elements or sequences used likely play an essential role in the genomic integration of this hybrid vector (3). Indeed, in our earlier study we demonstrated that a break occurred in the MoMLV element (5´ LTR) upstream of the luciferase cDNA during AdLTR-luc integration (3). The integration site in the host genome was random without any apparent preferential tendency (3). The specific mechanism by which AdLTR-luc can accomplish genomic integration is not yet understood. An important step to help understand the biological mechanism involved is to determine if integration also results in a break in the MoMLV element (3´ LTR) downstream of the luciferase cDNA. The purpose of the present study was to address this question. Our results clearly demonstrate that the integration event involved breaks in both MoMLV elements. Furthermore, our findings indicate that the DNA sequences between both MoMLV elements were integrated into genomic DNA without any adenoviral sequence.
Materials and Methods
Recombinant viral vectors
The replication-deficient recombinant adenoviral vector AdLTR-luc is based on the adenovirus serotype 5 (Ad5) genome and was constructed as previously reported (3). Briefly, E1 and E3 deletions were achieved by recombination of the pAC shuttle plasmid (a generous gift of Dr. C. Newgard) with pBHG10 (Microbix Biosystems Inc., Toronto, Ontario, Canada) (19). 2.7 kb of MoMLV sequence (which includes part of the envelope gene [1.5 kb], the 5´LTR [0.57 kb], and the packaging sequence [0.63 kb]) and 1 kb of MoMLV sequence (which contains ~ 0.5 kb of the envelope gene and an intact 3´LTR) from MoMLV were removed by EcoR I from the plasmid pXT1 (Stratagene, La Jolla, CA)(20). Not I linkers were added to both ends of the 2.7 kb of MoMLV sequence. Bam HI linkers were added to both ends of the 1 kb of MoMLV sequence. Fragments were ligated into pAC, with both MoMLV sequences placed in the deleted adenoviral E1 region, with the 2.7 kb MoMLV element upstream of the transgene and the 1 kb MoMLV element downstream. This construct did not contain any gag or pol sequences from MoMLV. The luciferase (luc) cDNA fragment was removed from the plasmid pGL2-Basic (Promega, Madison, WI) and ligated between the 2.7 kb of MoMLV sequence (5´end) and the 1 kb of MoMLV sequence (3´end). The luciferase gene was driven by the 5´LTR promoter. This plasmid was termed pACLTR-luc. The recombinant adenovirus (Fig. 1A), AdLTR-luc, was generated by homologous recombination of pACLTR-luc with pBHG10 in 293 cells (19).
Cell culture
The A5 epithelial cell line was derived from a rat submandibular gland (21) and grown in McCoy's 5A medium. A5 cells were transduced with AdLTR-luc at 50 pfu/cell. One week later the transduced A5 cells were cultured at very low cell density to form single clones on the bottom of a tissue culture dish. Isolated clones were harvested using a glass cylinder 0.5 cm in diameter. Luciferase activity was used to screen clones, and two luciferase positive clones (#10 and 11) were selected for further study (3). These two clones are the same clones reported in Zheng et al (2000). Clones #10 and 11 continue to express luciferase in vitro (Fig. 1B).
Luciferase assay
Cells were lysed in cell lysis buffer (Promega) for 15 minutes. Fifty microliters of the cell lysate were added to 100 µl of luciferase substrate, and light output was measured with a luminometer. Results are expressed as relative light units (RLU) per cell number.
Southern hybridization analyses
In this study, we mainly used restriction endonuclease digestion plus Southern hybridization to evaluate if there was a break in the MoMLV element downstream of the luciferase cDNA. Southern hybridization was used to detect expected DNA fragments. Thus, when the size of a DNA fragment was different from the expected size, it indicated that this part of the vector DNA was broken and integrated into the host genome. The genomic DNA used in the Southern hybridization analyses was extracted with a Non-Organic DNA Extraction kit (Intergen, Purchase, NY). Fifteen micrograms of genomic DNA from each sample were digested with different restriction endonucleases (GIBCO BRL, Rockville, MD; see Results) and separated on a 1% agarose gel. Nucleic acids were then transferred to nylon membranes. These blots were hybridized with an [α-32P] dCTP radiolabelled luciferase probe (a 540 bp Eco RI/Xba I fragment from the 5´end of the luciferase cDNA) and autoradiographed. In this study, the positive control samples used were from A5 cells two days after infection with AdLTR-luc. The negative control samples were obtained from uninfected A5 cells.
PCR assays
In this study, we also used a conventional PCR assay to evaluate if there was a break in the MoMLV element downstream of the luciferase cDNA. We hypothesized that if a specific fragment in AdLTR-luc was not amplified, when compared to a control sample, it would suggest that this part of MoMLV DNA was broken and integrated into the host genome. The genomic DNA used for the PCR assays was extracted with a Non-Organic DNA Extraction kit (Intergen). Positive DNA control samples were from A5 cells 2 days post transduction with AdLTR-luc. Genomic DNA from the two A5 transduced cell clones (#10 and 11) were from the same samples as used in Southern hybridizations. Negative controls used either water alone or genomic DNA from non-transduced A5 cells. PCR sensitivity was determined by using 0.5, 5, 50 and 500 ng AdLTR-luc positive control DNA as template. For individual PCR assays with genomic DNA from the transduced, cloned A5 cells, 0.5 µg, 3 µg and 10 µg template DNA were used. Taq DNA polymerase was obtained from GIBCO BRL (Rockville, MD). The primers 3´LTRf1 (5´-AAGAACAGATGGTCCCCAGATGCG-3´) and E2Bb1 (5´-AAGCCACGCCCACACATTTC-3´) produced the amplicon PCR 1 (1132 bp). The PCR 2 amplicon (786 bp) was amplified by 3´LTRf2 (5´-AACCCTCTTGCAGTTGCATCC-3´) and E2Bb2 (5´-GGAACGGGGTGTTTGACATGAC-3´). The PCR 3 amplicon (888 bp) was amplified by 3´LTRf2 and E2Bb1.
Results
Assessment of luciferase cDNA integrity
Initially, Southern hybridization with the luciferase probe was carried out on enzyme digested DNA samples from the two A5 cell clones studied, #10 and 11. Bam HI/Not I digestion of AdLTR-luc results in a 2.7 kb band that includes the entire luciferase cDNA (Fig. 1A). As shown in Fig. 1C and D, digested genomic DNA from both cloned cells had the same 2.7 kb band as the positive control, indicating the presence of an intact luciferase cDNA.
Structure of AdLTR-luc and Southern hybridization. A. Diagram of AdLTR-luc. AdLTR-luc contains the MoMLV elements described in the text: 2.7 kb upstream of the luciferase cDNA and 1 kb downstream. Luciferase served as a reporter gene, and the SV40 polyadenylation sequence was downstream of the luciferase cDNA. See text for details on construction. B. Luciferase activity from clones #10 and 11. C. Partial diagram of Bam HI and Not I target sites in AdLTR-luc. D. Southern hybridization to assess the integrity of the luciferase cDNA. DNA samples were obtained from cloned A5 cells (#s 10, 11) transduced with AdLTR-luc, or from non-transduced cells (N). Positive control (P) DNA was from uncloned cells transduced with AdLTR-luc 2 days before harvesting DNA. Each lane represents 15 µg of DNA applied and hybridized with the luciferase probe as described in the text. Note that cloned cell samples showed the same band (2.7 kb) as the positive control sample. At the left side of panel D, the migration positions of marker DNA (M; 1 Kb DNA ladder, GIBCO BRL, Rockville, MD) are shown in base pairs (bp).
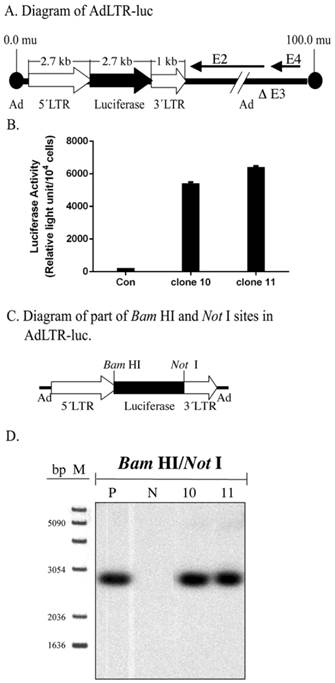
Determination of vector integration in A5 cell clones
To screen for AdLTR-luc integration, two restriction endonucleases, Xho I and Sal I, were used to digest DNA samples from the A5 cell clones. There are six Xho I sites, and three Sal I sites in AdLTR-luc. The first Xho I and Sal I sites from the 5´end of AdLTR-luc are located in the adenoviral E2 region (Fig. 2A). Therefore, if integration occurs at a location 5´to these sites, Southern hybridization would yield bands of different size from that seen with the positive control samples (smaller or larger than the positive control depending on Xho I and Sal I restriction endonuclease sites in the genome). Results in Fig. 2B show that both clones had hybridization-positive bands different in size compared to the positive control samples. Although these enzyme digestions showed that integration had occurred, the results could not clarify whether there was a break in the 2.7 kb MoMLV element and/or 1 kb MoMLV element in AdLTR-luc.
Southern hybridization to assess the presence of break points in either the 2.7 kb or 1 kb of MoMLV elements. All DNA samples were as in Fig. 1D. Samples were digested with Xho I, Sal I and and Nco I and hybridized with the luciferase probe. Panel A is a partial diagram of enzyme target sites in AdLTR-luc. Panel B shows Southern hybridization results. The migration positions of marker DNA (M) in base pairs (bp) are shown at the left.
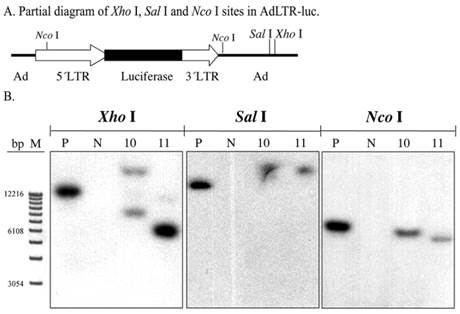
Assessment of possible break points in the MoMLV elements
To generally assess if break points were located in the 2.7 kb MoMLV element and/or 1 kb MoMLV element, Nco I was used to digest DNA samples. There is a Nco I site near the 3´end of the 1 kb of MoMLV sequence in the adenoviral genomic sequence. That Nco I site is 336 bp is downstream of the 3´end of the 1 kb MoMLV element. There is also a Nco I site at nt 647 of the 2.7 kb MoMLV element (Fig. 2A). If integration occurred, the Southern hybridizations of samples digested at these two restriction endonuclease sites would include hybridized bands of different size from that seen in the positive control. The results indicate that there is likely a break in either, or both, of the MoMLV elements of AdLTR-luc. As shown in Fig. 2B, both cell clones demonstrated band sizes different from the positive control samples.
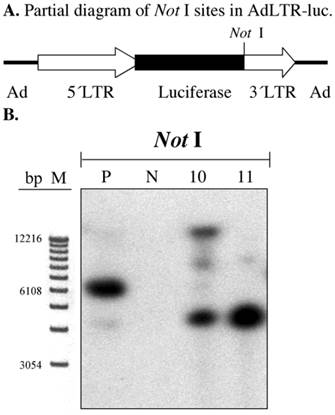
Demonstration of the break point in the 2.7 kb of MoMLV element
In our previous report we clearly demonstrated that there was a break in the 2.7 kb MoMLV element by a gene walking assay (3). We confirmed this result by Southern hybridization of A5 cell genomic DNA samples digested with Not I. The first Not I site encountered from the 5´end of AdLTR-luc is located between the luciferase cDNA and the 1 kb MoMLV element (Fig. 3A). There are no Not I sites in the 2.7 kb MoMLV element. There are only 454 bp of adenoviral sequence upstream of the 2.7 kb MoMLV element. Therefore, if the results of a Southern hybridization with the luciferase probe after digestion of genomic DNA with Not I indicate bands different in size from the positive control, integration must have occurred within the 2.7 kb MoMLV element. Results from Not I digested samples shown in Fig. 3B demonstrate that both A5 cell clones had different hybridization band sizes from the positive control sample.
Southern hybridization to assess the presence break points in the 2.7 kb MoMLV element. All samples were as in Fig. 2B. Samples were digested with Not I and hybridized with the luciferase probe. Panel A is a partial diagram of enzyme target sites in AdLTR-luc. Panel B shows Southern hybridization results. The migration positions of marker DNA (M) in base pairs (bp) are shown.
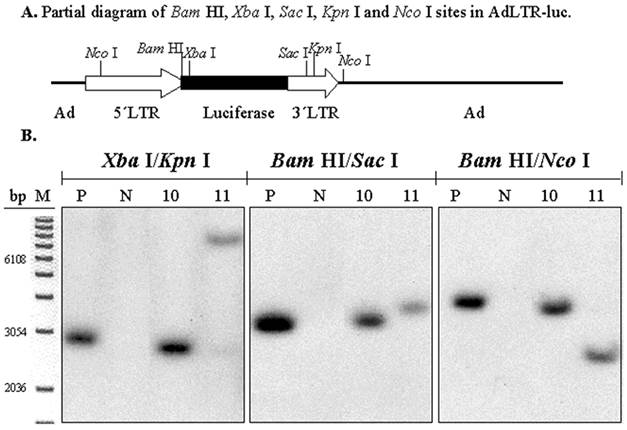
Assessment of possible break points in the 1 kb MoMLV element by Southern hybridization
To determine if there was also a break in the 1 kb MoMLV element, digestions with three pairs of restriction endonucleases were used (Fig. 4A). The experimental strategy was based on the presence of an intact luciferase cDNA, known from the results shown in Figs. 1C and D. There is a Xba I site in the luciferase cDNA, which is just upstream of the hybridization site of the luciferase probe. There is a Bam HI site between the 2.7 kb MoMLV element and the luciferase cDNA. There are Sac I and Kpn I sites in the 1 kb MoMLV element. Additionally, as shown in Fig. 2A, there are Nco I sites at nt 647 in the 5´MoMLV element and 336 bp from the 3´end of the 1 kb MoMLV element in the E2B adenoviral region (also see Fig. 4A). After digestion with either Xba I/Kpn I, Bam HI/Sac I or Bam HI/Nco I, clones # 10 and 11 exhibited different band sizes from positive control samples (Fig. 4B). The differences seen with clone # 11 are particularly clear with all digestions. Hybridization bands seen from Clone # 10 were slightly smaller than those of the positive control sample after Xba I/Kpn I and Bam HI/Nco I digestions, and slightly larger than that of the positive control samples after Bam HI/Sac I (Fig. 4B). The results with both clones #10 and 11 indicate that there is a break in the 1 kb MoMLV element of AdLTR-luc.
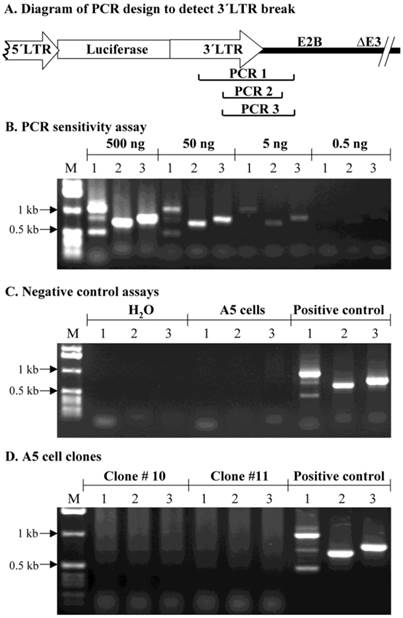
Assessment of possible break points in the 1 kb MoMLV element by PCR
As a separate approach to assess if the 1 kb MoMLV element was broken during integration, a PCR assay was used. PCR primers were synthesized to yield amplicons within the 1 kb MoMLV element and the E2B region of AdLTR-luc (Fig. 5A). Amplicons, PCR 1 (1132 bp), 2 (786 bp) and 3 (888 bp), were designed to amplify the junction between the 1 kb MoMLV element and E2B. An assay for PCR reaction sensitivity (Fig. 5B) showed that all three amplicons were clearly detectable when the template DNA from the AdLTR-luc positive control was decreased to 5 ng/reaction. However, when 0.5 ng template DNA per reaction was used, amplicons PCR 1, 2 and 3 were barely detected. Negative control experiments (Fig. 5C) showed that all three PCR amplicons could not be detected in the non-transduced A5 cell genomic DNA samples. The results in Fig. 5D show that amplicons, PCR 1, 2 and 3 could not be amplified even when ten µg template genomic DNA from clone #10 or 11 was present in each reaction (20-fold higher than maximum of positive control samples; also, two-thirds that used in Southern hybridization analyses). The failure to amplify PCR 1, 2 and 3 from clone #10 and 11 samples is consistent with the occurrence of a break in the 1 kb MoMLV element during integration.
Southern hybridization to determine the presence of break points in the 1 kb MoMLV element. All samples were as in Fig. 2B. Samples were digested with Xba I/Kpn I, Bam HI/Sac I or Bam HI/Nco I and hybridized with the luciferase probe. Panel A is a partial diagram of enzyme target sites in AdLTR-luc. Panel B shows Southern hybridization results. The migration positions of marker DNA (M) in base pairs (bp) are shown.
PCR assay to assess break points in the 1 kb MoMLV element. Panel A shows the design of PCR primers used to amplify the junction between the 3´LTR and the E2B region (PCR 1, 2 and 3) in AdLTR-luc. Panel B shows results of a PCR sensitivity assay. Panel C shows the two negative control assays used. For the non-transduced A5 cell genomic DNA samples, 0.5 µg DNA was used. Panel D shows results from cloned A5 cell samples (#10 and 11; 10 µg template DNA was used). Positive controls (P; 500 ng template AdLTR-luc DNA was used). The migration positions of marker DNA (M) in kilobase pairs (kb) are shown. The PCR assays used to detect amplicons 1, 2 and 3 are indicated by numbers 1, 2 and 3 in each corresponding lane of panels B - D.
Discussion
In the present study, we designed a series of Southern hybridization analyses to determine if the integration event seen following transduction of cells with AdLTR-luc involved breaks in both MoMLV elements upstream and downstream of the luciferase cDNA. Data from Bam HI/Not I digestions initially showed that both cell clones studied had an intact luciferase cDNA (Fig. 1D). Second, individual digestions with Xho I and Sal I showed that integration occurred in both A5 cell clones (Fig. 2B). These results were consistent with our previous findings (3). Results with Nco I digestions suggested that, indeed, there was a break in either (or both) the 2.7 kb MoMLV element or (and) the1 kb MoMLV element (Fig. 2B). Next, Not I digestion was used to show that there was a break in the 2.7 kb MoMLV element of AdLTR-luc in each cell clone (Fig. 3B). The demonstration of this break in the 2.7 kb MoMLV element was consistent with the gene walking data and the conclusion from our earlier report (3). We then used three pairs of restriction endonucleases (Xba I/Kpn I, Bam HI/Sac I, or Bam HI/Nco I) to digest cloned cell genomic DNA samples to test for a break point in the 1 kb MoMLV element (Fig. 4B). The results indeed indicated that a break in the 1 kb MoMLV element of AdLTR-luc also occurred. Finally, as a separate experimental approach to test for the apparent break point in the 1 kb MoMLV element, three sets of PCR primers were designed to amplify the junction between the 1 kb MoMLV element and the adenoviral E2B region (Fig. 5). Despite using as much as 10 µg genomic DNA in each PCR reaction, we were unable to detect any of the three amplicons in our assay. These data, although negative, further support the notion that there was a break within the 1 kb MoMLV element during the integration event. Thus, the demonstration of a break in the 1 kb MoMLV element in this report is novel, but in keeping with the important role of both LTRs in normal retroviral integration (11-18).
Clearly, AdLTR-luc retains some characteristics of MoMLV; AdLTR-luc integrates into the genome, and there are break points in both LTRs. However, classically, retroviral integration into the host cell's genome requires the 5´ and 3´LTRs along with virally encoded IN (11-18). While there is evidence for atypical integration in the absence of these classical requirements (22-24), AdLTR-luc integration without viral IN is particularly unusual. There was no retroviral contamination of, or generation in, the target cell lines used by us (not shown; tested by reverse transcriptase assays). Despite the absence of MoMLV IN, integration occurred with the LTR sequences apparently mediating the event. Cellular components can influence MoMLV integration (25-27), but there is no known mammalian protein with MoMLV-like IN activity. Our earlier studies showed that the break point localized in the 2.7 kb of MoMLV sequence (5´LTR in the 2.7 kb), is not at the classically recognized site (AATG) of MoMLV (11,14,15,17,22). Understanding the mechanism by which AdLTR-luc integration is accomplished will require a focus on both elements from MoMLV.
In summary, we have shown that AdLTR-luc achieved genomic integration and the break points for integration occurred within both the 2.7 kb MoMLV element and 1 kb MoMLV element. Clearly, both MoMLV elements play a major role in the integration event for this hybrid vector. Our previous report demonstrated that AdLTR-luc still retains the adenoviral characteristics of highly efficient transduction in vitro and in vivo in both dividing and non-dividing cells, while gaining the retroviral characteristics of genomic integration and long-term transgene expression. The present results provide an important biological characteristic of this hybrid vector and, thus, should facilitate further studies to understand AdLTR-luc's mechanism of integration.
Acknowledgements
The Division of Intramural Research, National Institute of Dental and Craniofacial Research, National Institutes of Health, provided all support for this research. We would like to thank Mrs. Beverly Handelman for helping to clone the A5 cells.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bilbao G, Feng M, Rancourt C, Jackson WH Jr, Curiel DT. Adenovi-ral/retroviral vector chimeras: a novel strategy to achieve high-efficiency stable transduction in vivo. FASEB J. 1997;11:624-34
2. Feng M, Jackson WH Jr Goldman CK, Rancourt C, Wang M, Dusing SK, Siegal G, Curiel DT. Stable in vivo gene transduction via a novel adenoviral/retroviral chimeric vector. Nat Biotechnol. 1997;15:866-70
3. Zheng C, Baum BJ, Iadarola MJ, O'Connell BC. Geneomic integration and gene expression by a modified adenoviral vector. Nature Biotech. 2000;18:176-180
4. Dales S, Chardonnet Y. Early events in the interaction of adenoviruses with HeLa cells. IV. Association with microtubules and the nuclear pore complex during vectorial movement of the inoculum. Virology. 1973;56:465-483
5. Greber UF, Suomalainen M, Stidwill RP, Boucke K, Ebersold MW, Helenius A. The role of the nuclear pore complex in adenovirus DNA entry. EMBO J. 1997;16:5998-6007
6. Greber UF, Webster P, Weber J, Helenius A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996;15:1766-1777
7. Harui A, Suzuki S, Kochanek S, Mitani K. Frequency and stability of chromosomal integration of adenovirus vectors. J. Virol. 1999;73:6141-6146
8. Schaack J, Ho WY, Freimuth P, Shenk T. Adenovirus terminal protein mediates both nuclear matrix association and efficient transcription of adenovirus DNA. Genes Dev. 1990;4:1197-1208
9. Varmus H.E. Mobile Genetic Elements. In: (ed.) Shapiro J. Retroviruses. New York: Academic Press. 1983:411-503
10. Craigie R, Fujiwara T, Bushman F. The protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell. 1990;62:829-837
11. Brown PO. Integration. In: (ed.) Coffin JM, Hughes SH, Varmus H.E. Retroviruses. Danvers: Cold Spring Harbor Laboratory Press. 1997:161-203
12. Asante-Appiah E, Skalka AM. Molecular mechanisms in retrovirus DNA integration. Antiviral Res. 1997;36:139-156
13. Donehower L, Varmus HE. A mutant murine leukemia virus with a single missense codon in pol is defective in a function affecting integration. Proc. Natl. Acad. Sci. USA. 1984;81:6461-6465
14. Goff S.P. Genetics of retroviral integration. Annu. Rev. Genet. 1992;26:527-544
15. Panganiban AT, Temin HM. The terminal nucleotides of retrovirus DNA are required for integration but not virus production. Nature. 1983;306:155-160
16. Panganiban AT, Temin HM. The retrovirus pol gene encodes a product required for DNA integration: identification of a retrovirus int locus. Proc. Natl. Acad. Sci. USA. 1984;81:7885-7889
17. Roth MJ, Schwartzberg PL, Goff SP. Structure of the termini of DNA intermediates in the integration of retroviral DNA: dependence on IN function and terminal DNA sequence. Cell. 1989;58:47-54
18. Schwartzberg P, Colicelli J, Goff S.P. Construction and analysis of deletion mutations in the pol gene of Moloney murine leukemia virus: a new viral function required for productive infection. Cell. 1984;37:1043-1052
19. Becker TC, Noel RJ, Coats WS, Gomez-Foix AM, Alam T, Gerard RD, Newgard CB. Use of recombinant adenovirus for metabolic engineering of mammalian cells. In: (ed.) M.G. Roth MG. Methods in Cell Biology. San Diego: Academic Press. 1994:161-189
20. Boulter CA, Wagner EF. A universal retroviral vector for efficient constitutive expression of exogenous genes. Nucleic Acids Res. 1987;15:7194
21. Brown AM, Rusnock EJ, Sciubba JJ, Baum BJ. Establishment and characterization of an epithelial cell line from the rat submandibular gland. J. Oral Pathol. Med. 1989;18:206-213
22. Colicelli J, Goff SP. Mutants and pseudorevertants of Moloney murine leukemia virus with alterations at the integration site. Cell. 1985;42:573-580
23. Hagino-Yamagishi K, Donehower LA, Varmus HE. Retroviral DNA integrated during infection by an integration-deficient mutant of murine leukemia virus is oligomeric. J. Virol. 1987;61:1964-1971
24. Murphy JE, Goff SP. A mutation at one end of Moloney murine leukemia virus DNA blocks cleavage of both ends by the viral integrase in vivo. J. Virol. 1992;66:5092-5095
25. Daniel R, Katz RA, Skalka AM. A role for DNA-PK in retroviral DNA integration. Science. 1999;284:644-647
26. Roe T, Chow SA, Brown PO. 3´-End processing and kinetics of 5´-end joining during retroviral integration in vivo. J. Virol. 1997;71:1334-1340
27. Lee MS, Craigie R. Protection of retroviral DNA from autointegration: Involvement of a cellular factor. Proc. Natl. Acad. Sci. USA. 1994;91:9823-9827
Author contact
![]() Corresponding author: Dr. Changyu Zheng, Bldg. 10, Room 1N113, MSC-1190, MPTB/NIDCR/NIH, 10 Center Drive, Bethesda, MD 20892-1190. Tel: (301)594-1924; Fax: (301)402-1228; Email: czhengnih.gov
Corresponding author: Dr. Changyu Zheng, Bldg. 10, Room 1N113, MSC-1190, MPTB/NIDCR/NIH, 10 Center Drive, Bethesda, MD 20892-1190. Tel: (301)594-1924; Fax: (301)402-1228; Email: czhengnih.gov
Received 2014-3-11
Accepted 2014-5-26
Published 2014-6-1