ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2014; 11(5):479-487. doi:10.7150/ijms.8340 This issue Cite
Research Paper
A Cell-Based Pharmacokinetics Assay for Evaluating Tubulin-Binding Drugs
Yuwei Wang1, Jihua Liu1, Jun Zhang1, Liping Wang1, Jonathon Chan1, Hai Wang1, Yi Jin1, Lei Yu1, David W. Grainger2, Wenbin Ying1 ![]()
1. Molecular Therapeutics Department, Nitto Denko Technical Corporation, 501 Via Del Monte, Oceanside, CA 92054, USA;
2. Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, UT 84112, USA.
Abstract
Increasing evidence reveals that traditional pharmacokinetics parameters based on plasma drug concentrations are insufficient to reliably demonstrate accurate pharmacological effects of drugs in target organs or cells in vivo. This underscores the increasing need to improve the types and qualities of cellular pharmacokinetic information for drug preclinical screening and clinical efficacy assessments. Here we report a whole cell-based method to assess drugs that disturb microtubule dynamics to better understand different formulation-mediated intracellular drug release profiles. As proof of concept for this approach, we compared the well-known taxane class of anti-microtubule drugs based on paclitaxel (PTX), including clinically familiar albumin nanoparticle-based Abraxane™, and a polymer nanoparticle-based degradable paclitaxel carrier, poly(L-glutamic acid)-paclitaxel conjugate (PGA-PTX, also known as CT-2103) versus control PTX. This in vitro cell-based evaluation of PTX efficacy includes determining the cellular kinetics of tubulin polymerization, relative populations of cells under G2 mitotic arrest, cell proliferation and total cell viability. For these taxane tubulin-binding compounds, the kinetics of cell microtubule stabilization directly correlate with G2 arrest and cell proliferation, reflecting the kinetics and amounts of intracellular PTX release. Each individual cell-based dose-response experiment correlates with published, key therapeutic parameters and taken together, provide a comprehensive understanding of drug intracellular pharmacokinetics at both cellular and molecular levels. This whole cell-based evaluating method is convenient, quantitative and cost-effective for evaluating new formulations designed to optimize cellular pharmacokinetics for drugs perturbing tubulin polymerization as well as assisting in explaining drug mechanisms of action at cellular levels.
Keywords: paclitaxel, whole cell analysis, in vitro drug testing, antimitotics, microtubule.
Introduction
Reliable drug absorption, distribution, metabolism and excretion (ADME) properties are essential for assessing pre-clinical prospects in drug development. Pharmacokinetics analysis is used to provide each step of ADME with quantitative information, including critical in vivo parameters for drug preclinical and clinical decision-making on therapeutic efficacy and index, and safety profiles. However, increasing reports reveal that traditional pharmacokinetics based on plasma drug concentrations do not correlate to drug pharmacological effects in target organs or cells since some drugs do not reach concentrations sufficient in their intracellular targets within the target cells to exert therapy.[1] Substantial uncertainty exists as to whether plasma drug concentrations consistently represent drug concentrations globally throughout target organs, or can ever actually reflect concentrations in specific target cells within these organs. To reach their intracellular targets, drug molecules must first penetrate the cell membrane, and may further require subsequent release from their carriers. The observed therapeutic effect is determined by the drug concentration at the (intracellular) binding sites and interactions between drug and intracellular targets.[2] Currently this information is extrapolated in vivo from dose-response studies using pharmacokinetic data and drug mass balancing models based on the drug's physical and chemical attributes in plasma. Such systemic in vivo analysis is unable to achieve a cellular focal point to describe a local therapeutic mechanism. Therefore, cellular pharmacokinetic studies can be a useful supplement to traditional in vivo clearance methods to assist traditional pharmacokinetics approaches in better predicting drug in vivo responses.
Among many drugs whose pharmacological responses rely closely on their cellular pharmacokinetics, the taxane tubulin-binding class of anticancer compounds is perhaps the most clinically impacting in current cancer chemotherapeutics.[3, 4] Paclitaxel (PTX), a central taxane therapeutic example, is a highly hydrophobic drug (log P ~ 3.96) and relatively low solubility (1 µg/ml). PTX exhibits significant anti-tumor activity by stabilizing cell microtubule dynamics, inducing sustained mitotic arrest and cell apoptosis in proliferating cells (i.e., tumor cells).[3] Due to its poor aqueous solubility, PTX is formulated using the excipient Cremaphor EL and ethanol (e.g., Taxol™), increasing its solubility some 6000-fold to improve its bioavailability.[5, 6] However, Cremaphor EL is associated with acute hypersensitivity reactions and other side effects (e.g., neuropathy).[7-9] Substantial effort has been expended to develop improved PTX formulations with improved bioavailability, biodistribution, and safety profiles. One area of active preclinical formulation has used nanoparticle drug carriers, primarily entrapped or encapsulated anti-cancer drugs in improved sub-micron vehicles and polymer-drug conjugates proposed to accumulate in tumors and inflamed tissues. Three successful clinical examples of PTX nanomedicines include albumin-entrapped PTX (Abraxane™), polymeric micelle formulation NK105, and the polymeric micelle formulation Genexol-PM™. All these carriers exhibit distinct PTX pharmacokinetic properties and therapeutic indices from PTX alone as free drug.[10]
For PTX formulations, drug efficacy depends on cell internalization and intracellular delivery, intracellular PTX release and interaction directly with cell microtubules in the cytosol. As shown by the contrast of the nanomedicine PTX formulations to free PTX,[10] in vivo plasma drug concentrations frequently do not reliably reflect pharmacological effects in target organs due to the failure to clearly distinguish systemic drug clearance by tumor uptake from the actual concentrated drug within diseased cells. This failure can result in surprising lack of efficacy in vivo and lack of correlation to in vitro cell toxicity profiles. For taxane tubulin-binding compounds, the kinetics of microtubule stabilization in cells directly reflect drug release rates from their formulations (e.g., vehicles, conjugates, particles).[11-13] Therefore, pharmacokinetic data documenting this effect are critical parameters for preclinical and clinical analysis of anti-tubulin drug candidates.
The direct pharmacological effect of anti-mitotic delivery is cell cycle arrest and induction of cell apoptosis.[14, 15] Accurate correlations between intracellular PTX drug release, cell tubulin assembly and cell viability provide reliable evidence of mechanism of drug action, facilitating ready lab-scale comparisons among drug candidates that perturb microtubulin assembly dynamics as their preferred mode of action. Thus, assays that directly correlate cellular tubulin dynamics to PTX efficacy in the complex cellular environment are attractive. However, analysis of intracellular drug release and drug pharmacology is complicated and difficult using traditional cell harvest, organic phase extraction and mass spectral analysis. Recently, a whole cell-based qualitative measurement of tubulin polymerization using α-tubulin staining was reported.[13] This method allows direct monitoring of tubulin assembly and quantitative analysis of tubulin polymerization in both time- and dose-dependent manners. We have adapted this approach to validate the utility of the assay for profiling the bioactivities of known anti-tubulin drugs. Abraxane® is a Cremophor-free, albumin-complexed PTX nanoparticle, diameter ~120 nm, developed as an alternative formulation to PTX (Taxol®) for treating metastatic breast cancer, permitting the administration of larger PTX doses while eliminating excipient side effects from Cremophor.[16, 17] Abraxane is frequently selected as a benchmark standard for evaluating other newly developed nanoparticle-based PTX formulations. Polymeric-drug conjugates are also developed to improve drug water-solubility, therapeutic index and tolerability of conventional taxanes.[18, 19] Plasma half-life of a poorly soluble drug can also be prolonged by its conjugation with water-soluble polymers that markedly improves solubility and frequently yields significant improvements in tumor concentrations.[16, 20, 21] Ideally, such polymer-drug conjugates should be inert during transport, and capable of releasing drug from the polymer carrier after selective endocytic uptake by tumor cells. The poly (L-glutamic acid)-PTX conjugate (PGA-PTX, also known as CT-2103) investigated in Phase I/II/III clinical trials,[22] was selected as a representative polymer-conjugated PTX for comparison. PGA-PTX has demonstrated complete regression of established tumors in murine models, and entered Phase I[23] and Phase II[24] clinical trials. However, in Phase III clinical trials, CT-2103 failed to show the expected enhanced efficacy compared to other non-nanoparticle standard PTX drugs against advanced non-small cell lung carcinoma.[25]
In this study, a cell-based in vitro assay system for analyzing intracellular PTX pharmacokinetics and efficacy using drug released at the cellular level compared Abraxane and PGA-PTX with control PTX. By analyzing intracellular tubulin polymerization kinetics, cell cycle G2 population arrest and cell viability in dose-response-time mode, an improved, comprehensive understanding of drug intracellular mechanism of action at a molecular level is obtained. The method has broader applicability for many other drug classes by adapting the assay format.
2. Materials and Methods
2.1 PTX formulations
PTX was purchased from NuBlocks (Vista, USA) and dissolved in DMSO at concentration of 5 µM. PGA was purchased (Sigma Chemical Co., USA) with relative average molecular weight of 18.98kDa. PGA-PTX was synthesized as reported [20] except using N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide instead of 1,3-dicyclohexylcarbodiimide as a drug coupling agent for ease of purification. The final product was characterized by proton NMR (JEOL Delta 400MHz). Particle size analysis for PGA-PTX used dynamic light scattering (Malvern Zetasizer Nano-ZS, Worcestershire, United Kingdom) at 2 mg/ml in pH 6.5 PBS-45% methanol media, and gel permeation chromatography (Wyatt GPC-MALS, Santa Barbara, USA). Levels of free PTX and 4-dimethylaminopyridine (DMAP, Sigma Chemical) in PGA-PTX were determined using SEC-HPLC (Agilent 1200, Shodex OHpak SB-804-HQ column with a dual-wavelength detector, detected at 254 nm and 228 nm, respectively). Commercially available Abraxane (Los Angeles, USA) was dissolved in PBS at 1 mg/ml. All other chemicals and reagents were obtained from Sigma Chemical Co. (St. Louis, USA), Invitrogen/Life Technologies, Inc. (Carlsbad, USA), Millipore Corporation (Temecula, USA), or Hyclone (Logan, USA).
2.2 Cell culture
HeLa cells and H460 cells (ATCC, USA) were grown in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 µg/ml penicillin and 100 U/ml streptomycin). Cells were incubated at 37oC in a 5% CO2/95% humidity atmosphere. All the cells used in the experiments were passaged at numbers less than 12.
2.3 Whole cell analysis of tubulin assembly and G2 arrest upon PTX dosing
Intracellular tubulin assembly level was evaluated using a published method with minor modifications.[13] In brief, HeLa cells were seeded at 100,000 cells/well in 24-well plates in RPMI-1640 (10% FBS, 100 µg/ml penicillin and 100 U/ml streptomycin) and treated with either Abraxane, PGA-PTX, or pure PTX at the indicated concentrations for different time durations. After PTX incubations, cells were trypsinized (trypsin-EDTA solution, Gibco, USA) for 5 min and re-suspended in 0.5 ml microtubule-stabilizing buffer (80 mM Pipes, 1 mM MgCl2, 5 mM EDTA, and 0.5% Triton X-100) containing 0.5% glutaraldehyde (Sigma) at pH 6.8. After 10 min. incubation at room temperature, glutaraldehyde was quenched by addition of 0.5 ml 0.25M glycine PBS solution. Cells were then spun at 1000g for 7 min and re-suspended in 20 µl of 50 µg/ml RNase A in Antibody Diluting Solution (AbDil: PBS, 0.2% Triton X-100, 2% bovine serum albumin, and 0.1% NaN3, pH 7.4) and incubated overnight at 4oC. The next day, cells were incubated with α-tubulin-FITC antibody (Sigma, 1:250) in AbDil for 3 hrs at room temperature, IgG1-FITC isotype from murine myeloma used as control (Sigma); further diluted in 200µl 50 µg/ml PI in FACS buffer (PBS, 2% BSA, 0.1% sodium azide, pH 7.2) followed immediately by flow cytometry analysis. The mean fluorescence intensity was recorded for each sample using a FACS C6 flow cytometer and analyzed using FlowJo software (Accuri). Cell cycle was identified based on propidium iodide (PI) staining.[13]
2.4 Intracellular PTX drug release assay
In order to confirm and compare intracellular PTX release results, the drug exposure assay was performed in two different cancer cell lines. Both HeLa cells and H460 cells were seeded in 6-well plates at 2x106 cells/well in serum-containing media. Cell confluence at the maximum time course evaluated was approximately 95%. Abraxane and PGA-PTX were dissolved in PBS, and PTX was dissolved in DMSO before application to cell cultures. Drugs were added at a final equivalent PTX concentration of 10 µg/ml in media and incubated for 19 hrs at 37oC. Culture medium was removed from each well, cells were harvested and lysed by adding 0.5 ml lysis buffer (Promega), and protein concentration was measured. Cell lysate (50 µL) was aliquoted and extracted with hexane/MTBE (1:3 v/v). Stable labeled internal standard PTX-d5 (IsoSciences, LLC, USA) was spiked before the addition of the extraction solvent and was used as the internal assay standard for free PTX. The organic phase including free PTX and PTX-d5 extract was collected and dried using a CentraVap (Labconco, USA) at 30°C. These dried extracts were reconstituted in 400µL of water/methanol (50:50 v/v) for free PTX analysis by HPLC/MS/MS (Agilent 1260 HPLC, Santa Clara, USA, a Leap HTC-xt Autosampler, Carrboro, USA, and AB SCIEX Qtrap 5500 mass spectrometer, Foster City, USA. A Phenomenex Kinetex C18 column (2.1x50, 2.6 µm) was used and maintained at 40oC, with 0.1% formic acid in water and 0.1% formic acid in methanol used as mobile phases A and B, respectively. Samples were eluted at 0.3mL/min with a 70% isocratic condition for free PTX and PTX-d5. Masses detected in ESI+ mode were 854.2/285.9 for free PTX, and 859.2/285.9 for PTX-d5. Data acquisition and processing was performed by Analyst software (AB SCIEX, USA). The methods were linear in the concentration range of 2 to 5,000 ng/mL for free PTX.[26]
2.5 In vitro cytotoxicity
Cells were seeded, cultured and treated with PTX-containing compounds as described above. Drug effects on cell proliferation and survival were evaluated (CellTiter 96® Aqueous One solution cell proliferation assay, Promega, USA). Drug dose-response curves were plotted for each condition after correction by background absorbance subtraction from controls. IC50 (50% drug inhibitory concentration) values were extrapolated from sigmoidal dose response curve fitting (GraphPad Prism software, San Diego, USA).
2.6 Statistical analysis
All experiments were independently repeated three times with multiple replicates within each run. Correlation coefficients (r) and statistical significance between cohorts in comparisons used Prism's correlation analysis based on two-tailed p-values (GraphPad Software, USA). Results were considered statistically significant if p < 0.01.
3. Results
3.1 Characterization of PGA-PTX polymer-drug conjugates
The average molecular weight of PGA-PTX was found to be 36.2 kDa by GPC-MALS, with polydispersity index (Mw/Mn) of 1.458. Conjugated PTX content was determined to be 20.6% by ultraviolet-visible optical absorbance methods. Conjugate particle size in mixed aqueous milieu determined using dynamic light scattering was found to be 23.44 nm in a solvent mixture containing 55 vol% 50 mM phosphate buffer (pH 6.5) and 45 vol% methanol.
3.2 Drug-cell correlations for tubulin assembly, cycle arrest and cell viability
3.2.1 Correlations between tubulin assembly and G2 population arrest and cell viability after 19-hour incubations with Abraxane in HeLa cells
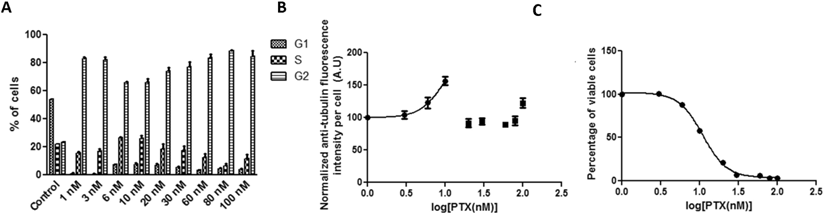
HeLa cells were treated with Abraxane in equivalent PTX concentrations in the media across 0-100 nM for 19 hours. Figure 1A shows that G2 arrest was progressively induced in a concentration-dependent manner, accompanying by a parallel decrease in the G1 population. Most of the cells were arrested in G2 phase when the PTX equivalent was equal or above 10 nM. The fluorescence which reflects cell tubulin assembly (Figure 1B) showed similar kinetics as the G2 cell population arrest results shown in Figure 1A, yielding a drug concentration value (potency) for inhibiting 50% tubulin assembly of 11.67 nM. The curve for tubulin fluorescence shifted to higher PTX concentration compared with G2 population arrest. The significant positive correlation between G2 cell population and tubulin assembly was shown in Figure 1D (r=0.8868). Specifically, when PTX concentration was equal to or above 20 nM, cell G2 populations and tubulin assembly were significantly correlated. The cytotoxicity assay results provided an IC50 value of 8.037 nM (Figure 1C), and cell viability showed significant negative correlation with tubulin assembly (Figure 1E, r=-0.9479).
3.2.2 Correlations between tubulin assembly and G2 population arrest and cell viability after 19-hour incubation with pure PTX in HeLa cells
In HeLa cell cultures, the kinetics of tubulin fluorescence intensity per cell and percentage of viable cells with increasing free PTX drug concentration were quite similar to that of Abraxane treatment (Figure 2). The drug concentration that yields 50% tubulin assembly increased to 11.29 nM. The cell IC50 of pure PTX treatment (13.4 nM) was higher than that of Abraxane. Cellular tubulin assembly was significantly positively correlated with G2 population (Figure 2D, r=0.9819). A strong negative correlation between tubulin assembly and cell viability was evident in Figure 2E, r=-0.9618.
3.2.3 Correlations between tubulin assembly and G2 population arrest and cell viability after 72-hour incubation with PGA-PTX in HeLa cells
No detectable differences in tubulin fluorescence intensity or cell viability were generated after incubating cultured cells with serial concentrations (up to 100 nM) of PGA-PTX after 19 hours. Therefore, the drug exposure duration was extended to 72 hours. After 72-hour incubations with PGA-PTX, cells were able to show similar kinetics (Figure 3) as the previous two PTX formulations. However, both tubulin assembly and cell viability curves for the polymer conjugate failed to reach the similar stationary phase in the sigmoidal curve. Cell G2 arrest and tubulin assembly significantly correlated with each other (Figure 3D, r=0.9842). The negative correlation between tubulin assembly and cell viability was significant as well (Figure 3E, r=-0.9075).
HeLa cells treated with Abraxane for 19 hours exposed to a series of increasing equivalent PTX concentrations. Cellular kinetics of (a) cell cycle arrest determined by PI staining and flow cytometry, (b) tubulin assembly assessed by anti-α-tubulin staining and flow cytometry, (c) cell proliferation measured by cytotoxicity assay, (d) correlations between tubulin assembly and G2 cell populations (r=0.8868, p=0.0014, R2=0.7863), and (e) correlations between tubulin assembly and cell proliferation (r=-0.9479, p=0.0001, R2=0.8986).
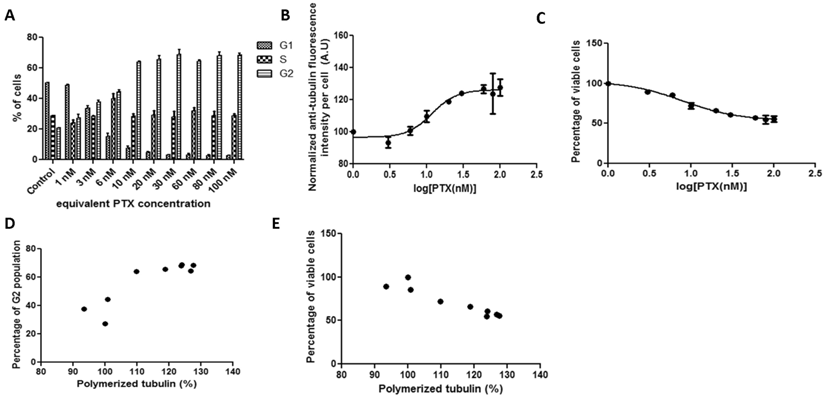
HeLa cells treated with pure PTX for 19 hours exposed to a series of increasing concentrations. Cellular kinetics of (a) cell cycle arrest determined by PI staining and flow cytometry, (b) tubulin assembly assessed by anti-α-tubulin staining and flow cytometry, (c) cell proliferation measured by cytotoxicity assay, (d) correlations between tubulin assembly and G2 cell populations (r=0.9819, p<0.0001, R2=0.964), and (e) correlations between tubulin assembly and cell proliferation (r=-0.9618, p=0.0005, R2=0.9251).
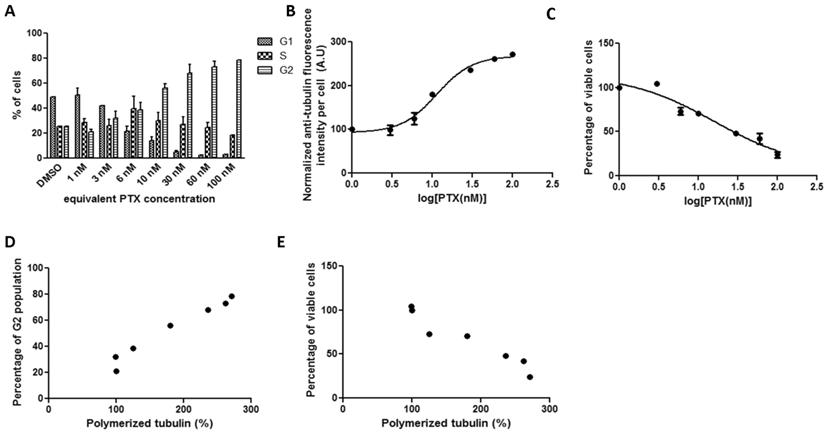
HeLa cells treated with PGA-PTX for 72 hours exposed to a series of increasing equivalent PTX concentrations. Cellular kinetics of (a) cell cycle arrest determined by PI staining and flow cytometry, (b) tubulin assembly assessed by anti-α-tubulin staining and flow cytometry, (c) cell proliferation measured by cytotoxicity assay, (d) correlations between tubulin assembly and G2 cell populations (r=0.9842, p<0.0001, R2=0.9686), and (e) correlations between tubulin assembly and cell proliferation (r=-0.9075, p=0.0048, R2=0.8235).
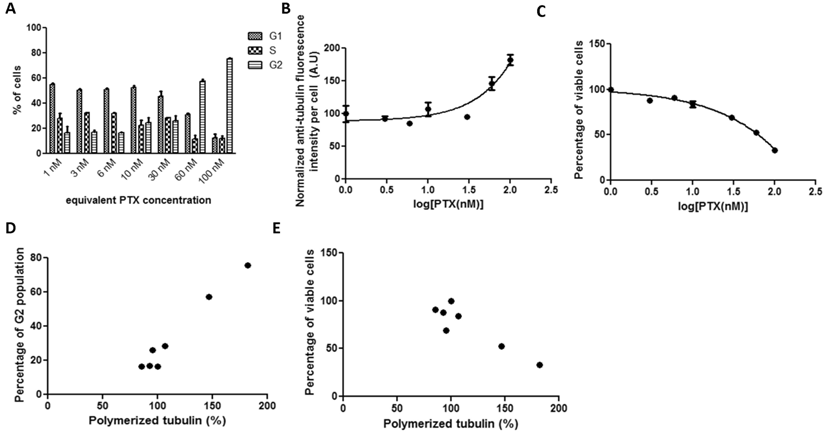
HeLa cells treated with Abraxane for 72 hours exposed to a series of increasing equivalent PTX concentrations. The kinetics of (a) cell cycle arrest determined by PI staining and flow cytometry, (b) tubulin assembly assessed by anti-α-tubulin staining and flow cytometry, and (c) cell proliferation measured by cytotoxicity assay.
3.2.4 Correlations between tubulin assembly and G2 population arrest and cell viability after 72-hour incubation with Abraxane in HeLa cells
In order to compare with PGA-PTX exposure, HeLa cells were incubated with Abraxane for 72 hours. Strong G2 arrest was achieved when equivalent PTX levels as low as 1 nM were dosed (Figure 4A). Anti-tubulin fluorescence intensity in the whole cell tubulin assembly assay dropped dramatically when the equivalent PTX concentration was equal to or above 20 nM, which may be attributed to its observed high cytotoxicity at this concentration as shown in Figure 4C. Cell viability versus drug concentration provided an IC50 of 10.99 nM. Correlations of G2 arrest and tubulin assembly were not possible in this case since sufficient data were not obtained.
3.3 Intracellular PTX release comparisons in cell cultures
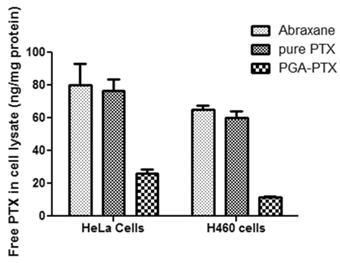
The traditional mass spectrometric methods (MS) for measurement of PTX in cell lysates have known intrinsic limitations. Therefore, two cell lines were tested for intracellular PTX amounts after 19 hours of incubation. Both cell lines showed the same MS results (Figure 5). Intracellular PTX amount (ng/mg protein) in the PGA-PTX treated group was significantly lower than both Abraxane and pure PTX treated cell groups. Free PTX amounts in the pure PTX treated group were slightly lower than that in Abraxane treated groups.
Comparison of the amount of intracellular PTX in HeLa and H460 cell cultures analyzed by MS after 19 hours of incubation with Abranane, PGA-PTX and pure PTX. Free PTX amounts (ng) were normalized to cellular protein amounts (mg) from cell lysates.
4. Discussion
In certain anticancer therapies, tubulin-binding compounds such as PTX are an unquestioned important clinical success. Additionally, tubulin binders have been actively investigated for treating diseases other than cancer.[27, 28] Improved formulations of tubulin-binding anti-tumor drugs are required to overcome current clinical limitations in their intrinsic toxicities and solubility, as well as bioavailability and efficacy. Development of optimized tubulin-binding drugs is therefore attractive and in some cases necessary for improved patient outcomes. Significantly, new means of reliably and rapidly assessing new lead compounds and formulations acting on cell tubulin dynamics are critical for efficient characterization and development in preclinical screens. However, traditional classical pharmacokinetic parameters based on in vivo plasma drug concentrations cannot sufficiently reflect local pharmacological effects in targeted diseased cells. Many new concepts in this regard involve improved drug carriers and delivery vehicles.[29, 30] In this regard, Abraxane was selected as a paclitaxel-based clinically familiar nanoparticle anti-cancer therapeutic and compared to polymer-drug conjugate-based nanoparticle PGA-PTX and pure PTX.
To detect intracellular released drug amounts, mass spectrometry is commonly used to measure free PTX in cell lysates. However, this method requires organic solvents to extract PTX from the aqueous lysate phase. It is difficult to differentiate actual released PTX from the cell-encapsulated but un-released PTX in delivered formulations. Both are indistinguishably extracted from cell lysate by organic solvent for assay. Since many new drug formulations are designed to optimize intracellular delivery and cellular pharmacokinetics, efficient in vitro cellular assay systems capable of analyzing intracellular interactions and specific drug release within whole cells would be beneficial, specifically for facilitating the development of tubulin-binding drugs. The cell-based analysis system presented here supports determination of intracellular drug pharmacokinetics based on correlating intracellular drug delivery, processing and release with their cell responses in vitro.
Regardless of various types of carriers used, drugs must first penetrate through the cell membrane, be released from the delivery vehicle, enter the cytosol at sufficient concentrations and bind to its intracellular target to exert their therapeutic effects. Consequently, intracellular drug trafficking and the amount of drug at specific target sites determine the observed pharmacological effects in the target organ.[2] PTX exerts its anticancer effects by promoting and stabilizing microtubule polymerization, blocking the cell cycle during mitosis and hence inducing apoptosis-programmed cell death.[29] In the experimental format described here, trace amounts of intracellular PTX could be correlated with amounts of polymerized tubulin or the percentage of cells arrested in G2 phase. Kinetics of intracellular PTX release could be magnified and represented as microtubule polymerization curves and cell G2 phase population curves. All of these parameters could be obtained in parallel based on this whole cell technique, thus cellular changes induced by drug dosing could be directly correlated with their cytotoxicity. Since these effects are achieved by the same PTX cell exposure, all parameters should correlate to support each other phenomenologically. By analyzing these correlations among three independent but unified biological phenomena, this cell-based assay system should support a drug dosing time-course within these cells.
In HeLa cell cultures incubated for 19 hours with both pure PTX and Abraxane, cells were able to generate sufficient differences in each parameter to yield significant dose-response curves, However, the polymer-based PGA-PTX nanoparticle failed to show these concentration and time-course differences, which implies a low level of released PTX inside the cells after 19 hours. Cellular effects induced by PTX, such as G2 arrest and tubulin assembly, must result from the combination of cell-mediated drug uptake and intracellular release. Abraxane, an albumin-bound nanoparticle of PTX, showed similar such kinetics as pure PTX; there is almost no delay of the onset of action for drug release from this nanoparticle formulation and it exhibits a lower IC50 than pure PTX. Regardless of the clinical route of administration, given equal PTX dosing in a micro-environment, tumor cells will show equal or better responses to Abraxane than to PTX alone. To date, the question of whether the Abraxane albumin nanoparticles dissolve or dose-dump their payload prior to cell uptake is unaddressed.[30] Some evidence has questioned if the proposed nanoparticle delivery method is the only mechanism for producing enhanced amounts of drug in tumor from Abraxane formulations.[31] As demonstrated in Figures 1 and 2, Abraxane particles are both taken up by cells and then release PTX intracellularly, or release free PTX outside cells that then diffuses into cells. Either way, it produces similar amounts of PTX intracellularly as PTX alone for the concentration range of 1 to 100 nM for 19 hours in culture. These data support the observation clinically that this particle-based therapeutic largely eliminates cell toxicities without compromising the therapeutic benefits of PTX. [17]
Using equivalent PTX amounts, PGA-PTX was not able to demonstrate these same effects until extending the experimental exposure to drug to 72 hours. Both cell tubulin assembly and G2 arrest kinetic curves did not reach their maximum saturation (i.e., the stationary phase in the sigmoidal curve), which implies a much slower PTX release than either Abraxane or PTX alone. In contrast, 72-hour cell treatment with Abraxane elicited significant cytotoxicity (Figure 4C) and significant G2 arrest was obtained even at 1 nM. Toxicity is too high to show gradual cell changes and correlations analogous to 19-hour cultures are not applicable. The slow release of PTX or the low intracellular amounts of PTX in PGA-PTX treated cells was proven using MS analysis in two different cell lines as shown in Figure 5. In PGA-PTX, PTX is conjugated with biodegradable poly-L-glutamic acid by an ester linkage between the γ-carboxylic acid moiety of glutamic acid and the PTX 2'-hydroxyl group.[32] PTX becomes active only after it dissociates from the polymeric backbone, since the conjugation site (2' hydroxyl of PTX) is crucial for tubulin binding and conjugated PTX is biologically inactive.[33] PTX release from this formulation in vitro occurs by (1) slowly hydrolysis from the polymer up to 14% after 14 hours; and (2) lysosomal protease release using enzymes such as cathepsin B degradation of the polyglutamic acid backbone after endocytic uptake, followed by hydrolysis of the PTX γ-carboxylate ester linkage.[34, 35] Results demonstrated in Figure 3 showed much slower PTX release from PGA-PTX compared to the other two formulations, indicating that even though these nanopolymer particles might accumulate at tumor sites in vivo, their slow PTX release could be sufficiently slow to limit therapeutic efficacy seen in human trials. Although clinical trials (phase I/II) of PGA-PTX (CT-2103) showed encouraging results in safety and efficacy, Phase III failed to show significant overall improvements in human efficacy in disease compared with other first-line therapies.[23-25, 36] As analyzed by this cell assay system that provides the same PTX amount/concentration equivalent to the same cell number, PGA-PTX released much less PTX to target sites inside cells due to the known slow polymer release of PTX both extracellularly or intracellularly.[32] This indicates much lower formulation potency compared with Abraxane or PTX alone. While the same amount of Abraxane and PGA-PTX can be delivered to the tumor site, Abraxane would show a much faster onset of action than PGA-PTX, perhaps reflecting its improved clinical performance compared to CT-2103.
5. Conclusions
The whole cell-based assay system described here for the taxane tubulin-binding drugs offers a new quantitative platform by which to assess, correlate and compare drugs that perturb microtubule dynamics on a cellular level. This cellular pharmacokinetics approach could be a supplement for traditional in vivo pharmacokinetics studies and should be useful for evaluating new drug carrier and delivery formulations designed for cell internalization where drug release kinetics are important. Deriving critical in vitro-in vivo relationships for drug action would be a primary benefit. In addition to preclinical and clinical analysis, this analysis system also could be used for observing drug mechanisms of action.
Nanoparticle therapeutics for anticancer purposes often requires that drug payloads be first delivered into tumor cells and then released into the cell cytoplasm. The complicated process of cellular delivery and intracellular release from delivery vehicles is difficult to evaluate accurately due to confounding influences from multiple intracellular factors. Therefore, a cell-based analysis system described here, including intracellular tubulin assembly determinations, cell cycle shifts, and cell proliferation, is useful for evaluating intracellular drug release and efficacy for taxane-based anticancer drugs. Each experiment in this system correlated with each other to affirm drug presence and bioactivity. Actually, this evaluation system can be used to assess all drug candidates that perturb microtubule dynamics in a cellular milieu. Taken together, the assay provides intracellular pharmacokinetic information to describe the intracellular drug dosing and better allow screening comparisons of novel nanoparticle delivery formulations to predict drug in vivo pharmacological effects.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48-58
2. Duvvuri M, Krise JP. Intracellular drug sequestration events associated with the emergence of multidrug resistance: a mechanistic review. Front Biosci. 2005;10:1499-509
3. Rowinsky EK. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu Rev Med. 1997;48:353-74
4. Goldspiel BR. Clinical overview of the taxanes. Pharmacotherapy. 1997;17:110S-25S
5. Fenner MH, Possinger K. Chemotherapy for breast cancer brain metastases. Onkologie. 2002;25:474-9
6. ten Tije AJ, Verweij J, Loos WJ, Sparreboom A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin Pharmacokinet. 2003;42:665-85
7. Rowinsky EK, Eisenhauer EA, Chaudhry V, Arbuck SG, Donehower RC. Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol. 1993;20:1-15
8. Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL. et al. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263-8
9. Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590-8
10. P. Lim Soo, J. Liu, C. Allen, H. Lee, M. Butler. Polymeric micelles for formulation of anti-cancer drugs. In: Amiji MM, editor. Nanotechnology for Cancer Drugs: CRC Boca Raton. 2007:317-46
11. Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci U S A. 1993;90:9552-6
12. Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol. 1998;10:123-30
13. Morrison KC, Hergenrother PJ. Whole cell microtubule analysis by flow cytometry. Anal Biochem. 2012;420:26-32
14. Adams JD, Flora KP, Goldspiel BR, Wilson JW, Arbuck SG, Finley R. Taxol: a history of pharmaceutical development and current pharmaceutical concerns. J Natl Cancer Inst Monogr. 1993:141-7
15. Rowinsky EK, Cazenave LA, Donehower RC. Taxol: a novel investigational antimicrotubule agent. J Natl Cancer Inst. 1990;82:1247-59
16. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653-64
17. Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A. et al. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res. 2005;11:4136-43
18. Alakhov V, Moskaleva E, Batrakova EV, Kabanov AV. Hypersensitization of multidrug resistant human ovarian carcinoma cells by pluronic P85 block copolymer. Bioconjug Chem. 1996;7:209-16
19. Minko T, Batrakova EV, Li S, Li Y, Pakunlu RI, Alakhov VY. et al. Pluronic block copolymers alter apoptotic signal transduction of doxorubicin in drug-resistant cancer cells. J Control Release. 2005;105:269-78
20. Li C, Yu DF, Newman RA, Cabral F, Stephens LC, Hunter N. et al. Complete regression of well-established tumors using a novel water-soluble poly(L-glutamic acid)-paclitaxel conjugate. Cancer Res. 1998;58:2404-9
21. Christie RJ, Grainger DW. Design strategies to improve soluble macromolecular delivery constructs. Adv Drug Deliv Rev. 2003;55:421-37
22. Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed Engl. 2006;45:1198-215
23. Veronese ML, Flaherty K, Kramer A, Konkle BA, Morgan M, Stevenson JP. et al. Phase I study of the novel taxane CT-2103 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2005;55:497-501
24. Sabbatini P, Aghajanian C, Dizon D, Anderson S, Dupont J, Brown JV. et al. Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. J Clin Oncol. 2004;22:4523-31
25. Jack W. Singer, Marc McKennon, Gabriella Pezzoni, Stefano di Giovine, Mara Cassin, Paola de Feudis, et al. Poly-L-Glutamic acid anti-cancer drug conjugates. In: L. Harivardhan Reddy PC, editor. Macromolecular Anticancer therapeutics. New York: Humana Press. 2010:154-6
26. Boddy AV, Plummer ER, Todd R, Sludden J, Griffin M, Robson L. et al. A phase I and pharmacokinetic study of paclitaxel poliglumex (XYOTAX), investigating both 3-weekly and 2-weekly schedules. Clin Cancer Res. 2005;11:7834-40
27. Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J. et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102:227-31
28. Burke WJ, Raghu G, Strong R. Taxol protects against calcium-mediated death of differentiated rat pheochromocytoma cells. Life Sci. 1994;55:313-9
29. Fauzee NJ. Taxanes: promising anti-cancer drugs. Asian Pac J Cancer Prev. 2011;12:837-51
30. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
31. Gardner ER, Dahut WL, Scripture CD, Jones J, Aragon-Ching JB, Desai N. et al. Randomized crossover pharmacokinetic study of solvent-based paclitaxel and nab-paclitaxel. Clin Cancer Res. 2008;14:4200-5
32. Shaffer SA, Baker-Lee C, Kennedy J, Lai MS, de Vries P, Buhler K. et al. In vitro and in vivo metabolism of paclitaxel poliglumex: identification of metabolites and active proteases. Cancer Chemother Pharmacol. 2007;59:537-48
33. Gueritte-Voegelein F, Guenard D, Lavelle F, Le Goff MT, Mangatal L, Potier P. Relationships between the structure of taxol analogues and their antimitotic activity. J Med Chem. 1991;34:992-8
34. Shaffer SA BLC, Kumar A, Singer JW. Proteolysis of XYOTAX by lysosomal cathepsin B; metabolic profiling in tumor cells using LC-MS. Eur J Cancer. 2002;38(Suppl. 7):s129
35. Singer JW, Baker B, De Vries P, Kumar A, Shaffer S, Vawter E. et al. Poly-(L)-glutamic acid-paclitaxel (CT-2103) [XYOTAX], a biodegradable polymeric drug conjugate: characterization, preclinical pharmacology, and preliminary clinical data. Adv Exp Med Biol. 2003;519:81-99
36. Langer CJ, O'Byrne KJ, Socinski MA, Mikhailov SM, Lesniewski-Kmak K, Smakal M. et al. Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin versus standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:623-30
Author contact
![]() Corresponding author: Wenbin Ying, MD.; Email: wenbin_yingnitto.co.jp.
Corresponding author: Wenbin Ying, MD.; Email: wenbin_yingnitto.co.jp.
Received 2013-12-11
Accepted 2014-2-27
Published 2014-3-18