ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2013; 10(12):1615-1624. doi:10.7150/ijms.6635 This issue Cite
Research Paper
Interaction of RAS Activation and Lipid Disorders Accelerates the Progression of Glomerulosclerosis
Kun-Ling Ma1, Jie Ni1, Chang-Xian Wang2, Jing Liu1, Yang Zhang1, Yu Wu1, Lin-Li Lv1, Xiong-Zhong Ruan3, Bi-Cheng Liu1 ![]()
1. Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, Nanjing City, Jiangsu Province, P.R. China.
2. Department of Infection Management, Zhong Da Hospital, Southeast University School of Medicine, Nanjing City, Jiangsu Province, P.R. China.
3. Centre for Nephrology, University College London (UCL) Medical School, Royal Free Campus, UK.
Abstract
Background: The activation of the renin-angiotensin system (RAS) and lipid disorders are major risk factors in progressive chronic kidney disease. This study aimed to investigate the potential synergistic mechanisms of RAS activation and lipid disorders that contribute to glomerulosclerosis. Materials and methods: Human renal mesangial cells (HMCs) were treated with 10-7 mol/L angiotensin II (Ang II) or with 30 μg/ml cholesterol and 1 μg/ml 25-hydroxycholesterol (lipid loading) for 24 hours. Lipid accumulation in the cells was evaluated by Oil Red O staining and intracellular cholesterol quantitative assays. The gene and protein expression of molecules in the low-density lipoprotein receptor (LDLr) pathway, the RAS family, and the extracellular matrix were examined by real-time polymerase chain reaction and Western blotting. The translocation of sterol regulatory element-binding protein (SREBP) cleavage activating protein (SCAP), which escorts SREBP-2 from the endoplasmic reticulum (ER) to the Golgi, was examined by immunofluorescent staining. Results: Ang II increased lipid droplet accumulation in HMCs. Further analysis revealed that Ang II increased the mRNA and protein expression of LDLr, SCAP, and SREBP-2. This increase was correlated with an enhanced translocation of the SCAP/SREBP-2 complex from the ER to the Golgi in HMCs that was induced by Ang II, thereby activating LDLr gene transcription. Interestingly, lipid loading increased the mRNA and protein expression of angiotensinogen, Ang II, renin, angiotensin-converting enzyme, angiotensin II type 1 receptor, and type 2 receptor in HMCs with increased mRNA and protein expression of collagen I, α-smooth muscle actin, and fibronectin. Conclusions: This study demonstrates that the interaction of RAS activation and lipid disorders accelerates the progression of glomerulosclerosis.
Keywords: renin-angiotensin system, lipid disorder, glomerulosclerosis, low-density lipoprotein receptor, extracellular matrix
Introduction
Glomerulosclerosis, which is characterized by an excessive deposition of extracellular matrix (ECM) in the mesangial area (1), is a final common pathway leading to the loss of renal function in a variety of chronic kidney diseases (CKD). Changes in the amount and composition of the ECM from the glomerular mesangium include the expression of type I collagen and other types of collagen, which are normally limited to the interstitium, the upregulation of α-smooth muscle actin (α-SMA), and the accumulation of plasma-type fibronectin (2). Moreover, the ECM is an important regulator of cell phenotype, including cell survival, for a wide variety of cell types, such as glomerular mesangial cells (3), suggesting that the development of glomerulosclerosis may reflect the disruption of cellular signaling that is secondary to the altered composition of the ECM.
It is known that both the maladaptive activation of the renin-angiotensin system (RAS) and dyslipidemia play deleterious roles in progressive kidney damage. Angiotensin II (Ang II) is produced as a result of RAS activation and has important nonhemodynamic effects that have been implicated in the pathogenesis of glomerulosclerosis by promoting mesangial cell proliferation and increasing ECM deposition (4). Meanwhile, there is growing evidence that abnormal lipid metabolism and the renal accumulation of lipids play crucial roles in the pathogenesis of renal disease (5). Reduced clearance and increased plasma levels of small, dense low density lipoprotein (LDL) particles facilitate their entrance into leukocytes, macrophages, and arterial walls, in which the accelerated oxidation of these particles causes renal and vascular damage due to the production of inflammatory mediators and reactive oxygen species. However, the exact mechanism by which RAS activation interacts with lipid disorders in glomerulosclerosis is unknown.
There is increasing evidence that there is cross-talk between dyslipidemia and RAS activation in atherogenesis (6, 7). Catar et al. has reported that native or oxidized LDL enhances the expression levels of angiotensin-converting enzyme (ACE) and Ang II type 1 receptor (AT1) in human endothelial cells through LDL receptors and scavenger receptors (8). Meanwhile, Ang II facilitates the oxidation of LDL and its uptake by vascular smooth muscle cells and macrophages (9). Because mesangial cells and vascular smooth muscle cells share a common embryonic origin and many other features, the interaction between dyslipidemia and RAS activation in atherosclerosis may provide insight into the mechanisms that lead to glomerulosclerosis. Therefore, this study aimed to investigate the potential synergistic mechanisms of RAS activation and lipid disorders that affect the progression of glomerulosclerosis in human renal mesangial cells (HMCs).
Materials and methods
Cell culture
An established stable human mesangial cell line (kindly provided by Prof. Ruan from the Centre for Nephrology, University College London Medical School, UK) was used in all experiments. HMCs were immortalized by the SV-40 transfection of the H-Ras oncogene and maintained their basic biological features. The cells were cultured in RPMI 1640 (Gibco, USA) containing 1% penicillin and streptomycin (Invitrogen, USA), 2 mmol/L L-glutamine (Sigma, USA), and 10% heat-inactivated fetal calf serum (Gibco, USA). The cells were maintained in an incubator with 5% CO2 at 37ºC. At 70-80% confluence, the cells were synchronized with a serum-free culture medium containing 0.2% fatty acid-free bovine serum albumin (BSA, Gibco, USA) for 24 hours and subsequently stimulated with 30 μg/ml cholesterol (Sigma, USA) and 1 μg/ml 25-hydroxycholesterol (Sigma, USA) or with 10-7 mol/L angiotensin II (Ang II, Sigma, USA) alone for 24 hours.
MTT reduction assay
A methylthiazoletetrazolium (MTT) reduction assay was used as a quantitative index of cell viability. Each experiment was typically performed with 5 separate wells of HMCs in 96-well plates under identical conditions. After incubation with the compounds listed above for 24 hours, 20 μl of MTT (5 mg/ml, Invitrogen, USA) was added, and the cells were cultured for an additional 4 hours. Subsequently, the cells were lysed using dimethylsulfoxide (150 μl/well). When the formazan crystals were completely dissolved, the optical density (OD) was measured at 490 nm with a Microplate Reader Model 3550-UV Spectrophotometer (BioRad Laboratories, France).
Cell cycle analysis
Cell cycle analysis was performed using flow cytometry. After 24 hours of treatment with different compounds, the cells were harvested, fixed in cold 70% (vol/vol) ethanol, and stored at -4°C. The cells were then washed twice with cold phosphate-buffered saline (PBS) and incubated in 500 μl of propidium iodide/RNase staining buffer (BD Biosciences, USA) at 37°C for 1.5 hours in the dark. Each sample was then analyzed using a Coulter Epics XL Flow Cytometer (Miami, USA), and the percentage of cells in the G1, S, and G2/M phases of the cell cycle was determined.
Morphological examination
Lipid accumulation in the HMCs was evaluated by Oil Red O staining. Briefly, the cells were plated in 12-well (Corning, USA) plates and incubated in serum-free RPMI 1640 with or without Ang II. After 24 hours, the cells were washed three times with PBS, fixed for 30 minutes with a 5% formalin solution in PBS, stained with Oil Red O (Sigma, USA) for 30 minutes, and counterstained with hematoxylin (Sigma, USA) for 5 minutes. Finally, the cells were examined by light microscopy.
Quantitative measurements of intracellular free cholesterol/cholesterol ester
Quantitative measurements of intracellular total and free cholesterol were analyzed using the method described by Gamble et al. (10) Briefly, cells were collected and lipids were extracted by adding 1 mL of chloroform/methanol (2:1). After sonification, the samples were centrifuged, and the lipid phase was collected. The samples were dried in vacuum and were then dissolved in 2-propanol containing 10% Triton X-100. Cholesterol ester was converted to free cholesterol by cholesterol ester hydrolase (Sigma, USA) to determine the amount of total cholesterol. The concentration of total and free cholesterol per sample was analyzed using a standard curve and normalized against total cell protein. The concentration of cholesterol ester was calculated by subtracting the amount of free cholesterol from the total cholesterol.
Confocal microscopy
For confocal microscopy, the HMCs cultured in a glass bottom dish (Corning, USA) were washed, fixed, and permeabilized. The cells were then incubated with goat anti-human SCAP antibody (1:100; Santa Cruz, USA) and rabbit anti-human Golgi antibody (1:100; Santa Cruz, USA), followed by secondary fluorescent antibodies (donkey anti-goat Alexa Fluor 488 for SCAP and donkey anti-rabbit Alexa Fluor 594 for Golgi). After washing, the cells were examined by laser confocal microscopy (Olympus, Japan). The colocalization efficiency of SCAP with Golgi was quantified by the software of Image-Pro Plus version 6.0.
Real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted from the HMCs, and reverse transcription was performed using RNAiso Plus (Takara, Japan) in accordance with the manufacturer's protocols. RT-PCR was performed on an ABI PRISM 7300 real-time PCR System (Applied Biosystems, USA) using SYBR Green dye. The primers used for RT-PCR are given in Table 1. β-actin served as an internal reference gene. The results were analyzed using Sequence Detection Software version 1.4 (Applied Biosystems, USA). The relative gene expression of each target gene was quantified against a standard curve. The pre-PCR product of each gene was used as the standard, and the standard curve was established with a 10-fold serial dilution of the product. The standard curve was included in all PCR runs. The expression levels of target genes of interest were normalized against the expression level of a reference gene abundance. All measurements were performed in duplicate.
The primers for real-time PCR
| Gene | Primer sequence |
|---|---|
| Collagen I | 5'-GGCTTCCCTGGTCTTCCTGG-3'-sense 5'-CCAGGGGGTCCAGCCAAT-3'-antisense |
| α-SMA | 5'-GACAATGGCTCTGGGCTCTGTAA-3'-sense 5'-ATGCCATGTTCTATCGGGTACTTCA-3'-antisense |
| Fibronectin | 5'-GAGCTGCACATGTCTTGGGAAC-3'-sense 5'-GGAGCAAATGGCACCGAGATA-3'-antisense |
| LDLr | 5'-GTGTCACAGCGGCGAATG-3'-sense 5'-CGCACTCTTTGATGGGTTCA-3'-antisense |
| SCAP | 5'-GGGAACTTCTGGCAGAATGACT-3'-sense 5'-CTGGTGGATGGTCCCAATG-3'-antisense |
| SREBP-2 | 5'-CCGCCTGTTCCGATGTACAC-3'-sense 5'-TGCACATTCAGCCAGGTTCA-3'-antisense |
| Angiotensinogen | 5'-GATGTTGCTGCTGAGAAGATTG-3'-sense 5'-GGAAGTGGACGTAGGTGTTGA-3'-antisense |
| Renin | 5'-GAGGCTGACACTTGGCAACA-3'-sense 5'-CGCCATAGTACTGGGTGTCCAT-3'-antisense |
| ACE | 5'-CACTATCAAGCGGATCATAAAGAAG-3'-sense 5'-CACGCTGTAGGTGGTTTCCATA-3'-antisense |
| AT1 | 5'-ACCTGGCTATTGTTCACCCAAT-3'-sense 5'-TGCAGGTGACTTTGGCTACAAG-3'-antisense |
| AT2 | 5'-CCACCCTTGCCACTACTAGCA-3'-sense 5'-ATTGTTGCCAGAGATGTTCACAA-3'-antisense |
| β-actin | 5'-AAAGACCTGTACGCCAACAC-3'-sense 5'-GTCATACTCCTGCTTGCTGAT-3'-antisense |
Western blotting
Identical amounts of total protein extracted from the HMCs were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred onto PVDF membranes. The membranes were then blocked with 5% skimmed milk in Tris-buffered saline with 0.5% Tween 20 for 1 hour at room temperature. The membranes were subjected to Western blotting using anti-human polyclonal antibodies against ACE, AT1, AT2, renin, fibronectin, LDLr, SREBP-2, SCAP, Golgi (Santa Cruz, USA), collagen I, α-SMA (Abcam, USA), angiotensinogen (Abbiotec, USA), and Ang II (Novus bio, USA) overnight at 4ºC, followed by horseradish peroxidase-labeled secondary antibodies for 1 hour at room temperature. β-actin was used as an internal sample loading control and was detected with a mouse monoclonal antibody (Santa Cruz, USA). Signals were detected using an ECL advanced system (GE Healthcare, UK). Relative expression levels were determined by normalization against β-actin.
Statistical analysis
All data were expressed as the mean ± the standard error of the mean (SEM). The Student's t-test was used to analyze the statistical significance between two groups and one-way analysis of variance (ANOVA) was used for more than two groups before expressing the results as a percentage of the control using the SPSS13.0 statistical software. P < 0.05 was considered to be statistically significant.
Results
Ang II increased lipid accumulation in HMCs
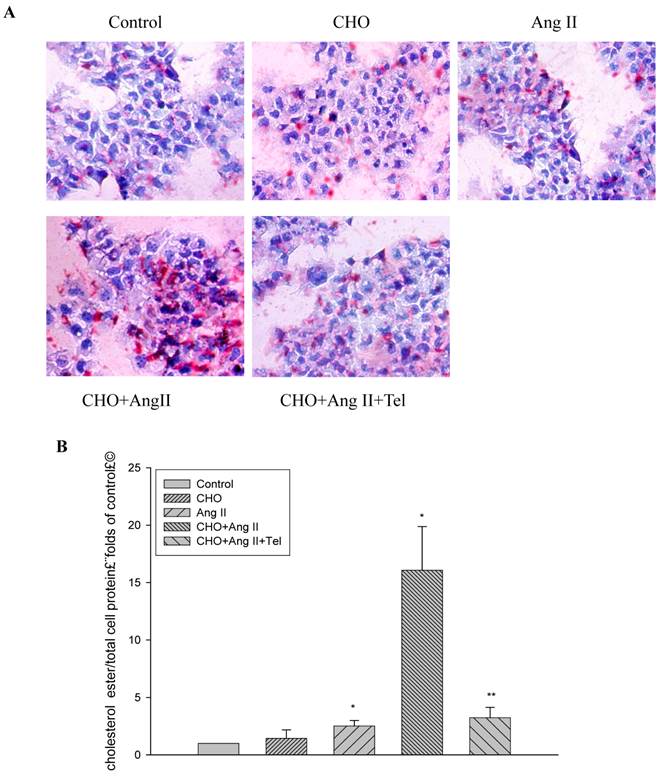
Using Oil Red O staining, we checked the morphological changes of the HMCs treated with or without Ang II (10-7 mol/L) for 24 hours. The results showed that Ang II significantly increased lipid droplet accumulation in HMCs (Fig. 1A). Furthermore, quantitative intracellular cholesterol analysis confirmed that Ang II induced cholesterol ester accumulation in HMCs (Fig. 1B). However, these effects were inhibited by telmisartan (Fig.1A and 1B).
Visualization of lipid accumulation in HMCs treated with Ang II. HMCs were incubated for 24 hours in a serum-free medium in the absence (Control) or presence of 30 μg/mL cholesterol (CHO), or 10-7 mol/L Ang II (Ang II), or 30 μg/mL cholesterol + 10-7 mol/L Ang II (CHO+Ang II), or 30 μg/mL cholesterol + 10-7 mol/L Ang II + 10-6 mol/L telmisartan (CHO+Ang II+Tel). (A) The cells were examined for lipid accumulation by Oil Red O staining. The results are typical of those observed in four separate experiments (magnification ×200). (B) Effects of Ang II on intracellular free cholesterol and cholesterol ester in HMCs. Intracellular free cholesterol and cholesterol ester were assayed as described in Materials and Methods. Values are expressed as the mean ± SD of triplicate wells from four experiments. *P<0.05 vs. control, P<0.01 vs CHO+Ang II.
Ang II disrupted LDLr feedback regulation in HMCs
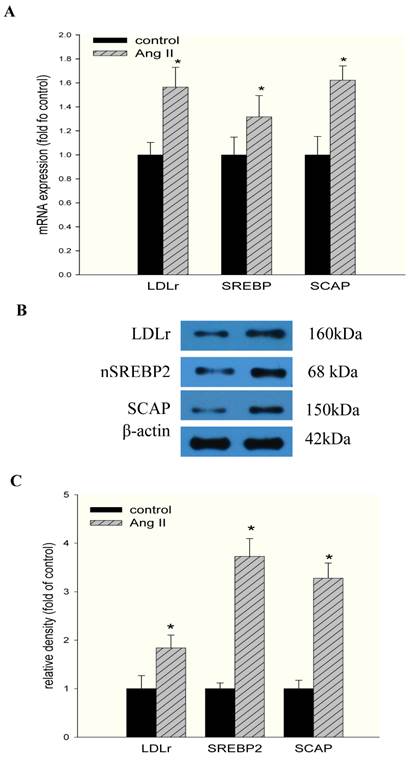
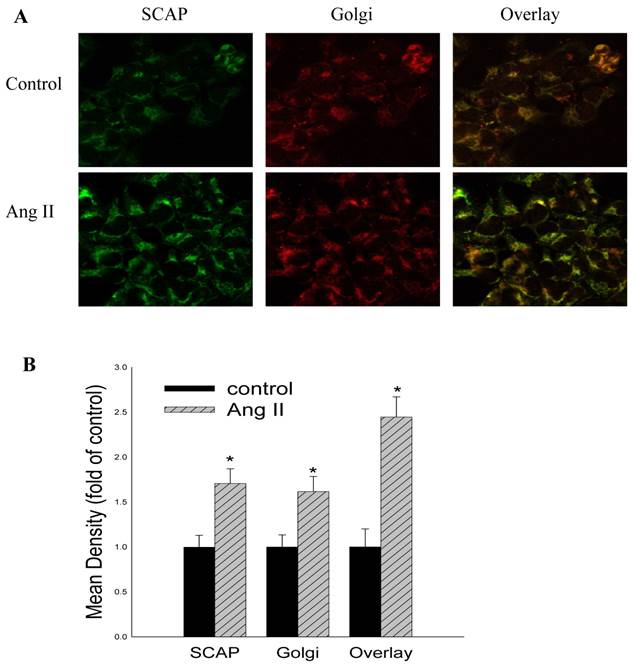
To gain further insight into the mechanisms regulating these phenomena, we studied the effects of Ang II on the LDLr pathway, which mediates cholesterol uptake. Ang II significantly upregulated LDLr, SREBP-2, and SCAP gene and protein expression in HMCs, as demonstrated by RT-PCR (Fig 2A) and Western blotting (Fig 2B and Fig 2C). Using confocal immunofluorescent staining, we further investigated the effects of Ang II on the translocation of SCAP escorting SREBP-2 from the ER to the Golgi in HMCs. The results demonstrated that Ang II increased SCAP translocation from the ER to the Golgi in HMCs compared with the control (Fig. 3A and 3B).
Lipid loading increased the expression of RAS components in HMCs
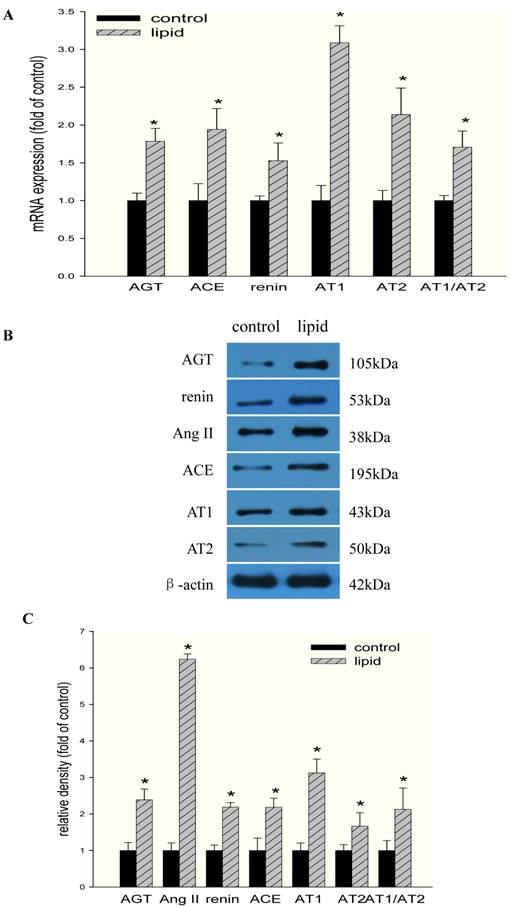
We then investigated the effects of lipid loading on the expression of RAS components in HMCs. As shown in Fig 4A, lipid loading significantly increased the mRNA expression of angiotensinogen, renin, ACE, AT1, and AT2 in HMCs. AT1/AT2 also increased after lipid loading, suggesting that the balance between the expression of these two receptors was disrupted and that the elevation of AT1 expression may play a more central role in this process. These results were further confirmed at the protein level by Western blotting (Fig. 4B and Fig. 4C).
Lipid loading modified the phenotype and function of HMCs
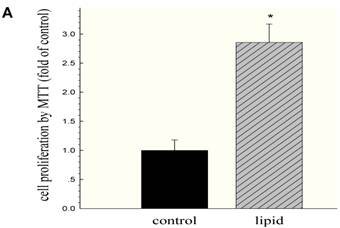
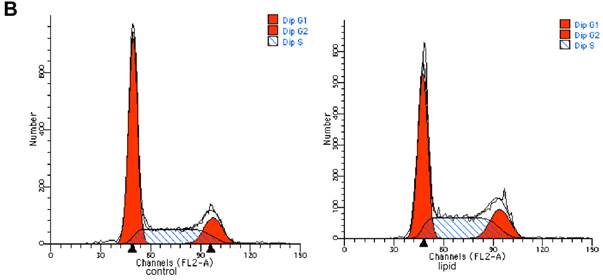
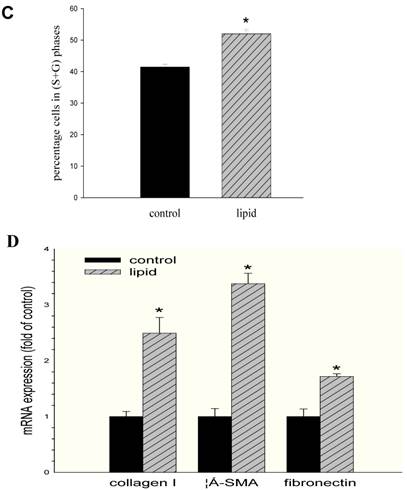
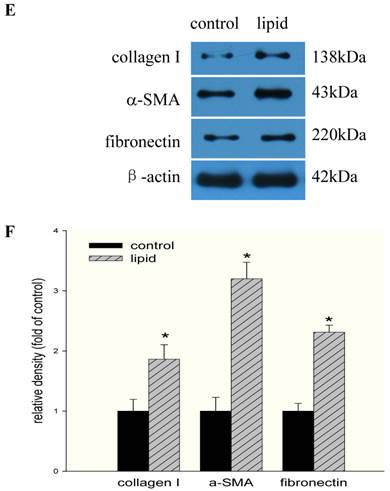
We further evaluated the effects of lipid loading on the phenotype and function of HMCs. The results showed that lipid loading induced HMC proliferation after 24 hours (Fig. 5A). Flow cytometry showed that lipid loading increased the percentage of cells in the active division phases (S and G2 phases), consistent with the findings from the MTT reduction assay (Fig. 5B and Fig. 5C). We then checked the effects of lipid loading on the expression of extracellular matrix proteins in HMCs. As shown in Fig. 5D-5F, there was increased expression of collagen I, α-SMA, and fibronectin in the lipid loading group compared with the control group at the mRNA level and protein level, suggesting that lipid loading may mediate ECM excretion by HMCs.
Effects of Ang II on the mRNA and protein expression of LDLr, SCAP, and SREBP-2 in HMCs. Cells were incubated in a serum-free medium (control) or a serum-free medium with 10-7 mol/L Ang II (Ang II group) for 24 hours. (A) The mRNA expression of LDLr, SCAP, and SREBP-2 was determined by real-time PCR. β-actin served as the housekeeping gene. The results represent the mean ± SD from 4 experiments. (B and C). The protein expression of LDLr, SCAP, and SREBP-2 was determined by Western blotting. The histogram represents the mean ± SD of the densitometric scans for the protein bands from four experiments normalized against β-actin and expressed as a percentage of the control.
Effects of Ang II on the translocation of SCAP escorting SREBP-2 from the ER to the Golgi in HMCs. HMCs were incubated in a serum-free medium (control) or a serum-free medium with 10-7 mol/L Ang II (Ang II group) for 24 hours. HMCs in chamber slides were then washed, fixed, and permeabilized. The cells were then incubated with a rabbit anti-human SCAP antibody (1:200 dilution) and a mouse anti-human Golgi antibody (1:200 dilution), followed by secondary fluorescent antibodies (goat anti-rabbit Fluor 488 for SCAP and goat anti-mouse Fluor 594 for Golgi). After washing, the cells were examined by confocal microscopy (A). The colocalization efficiency of SCAP with Golgi was quantified and expressed as Mean ± SD (×400). (B). * p <0.05 vs control.
Effects of lipid loading on the expression of RAS components in HMCs. HMCs were incubated in a serum-free medium for 24 hours and then maintained in a serum-free medium (control) or a serum-free medium containing 30 μg/ml cholesterol and 1 μg/ml 25-hydroxycholesterol (lipid group) for 24 hours. (A) Real-time PCR for the mRNA expression of RAS components (angiotensinogen, renin, ACE, AT1, and AT2) in HMCs with or without lipid treatment. AT1/AT2 was evaluated, and β-actin served as the housekeeping gene. The results represent the mean ± SD from four experiments. (B and C) Western blotting analysis of the protein expression of RAS components (angiotensinogen, Ang II, renin, ACE, AT1, and AT2). The histogram represents the mean ± SD of the densitometric scans for the protein bands of RAS components from four experiments normalized against β-actin. *P<0.05 vs. control.
Effects of lipid loading on the phenotype and function of HMCs. HMCs were incubated in a serum-free medium for 24 hours and then maintained in a serum-free medium (control) or a serum-free medium containing 30 μg/ml cholesterol and 1 μg/ml 25-hydroxycholesterol (lipid group) for 24 hours. (A) Effects of lipid loading on HMC proliferation, as assessed by a MTT reduction assay. (B and C) Effects of lipid loading on the distribution of cell cycle in HMCs, as examined by flow cytometry. (D) Real-time PCR for the mRNA expression of collagen I, α-SMA, and fibronectin in HMCs with or without lipid treatment. β-actin served as the housekeeping gene. The results represent the mean ± SD from four experiments. (E and F) Western blotting analysis for the protein expression of collagen I, α-SMA, and fibronectin. The histogram represents the mean ± SD of the densitometric scans for the protein bands from four experiments normalized against β-actin. *P<0.05 vs. control.
Discussion
Some studies have reported that RAS activation combined with dyslipidemia plays an important role in the development of glomerulosclerosis (11, 12). In CKD patients also suffering from hypertension and hyperlipidemia, there is more severe damage and accelerated sclerosis in the glomeruli (13). Using apolipoprotein E knockout mice, our previous study demonstrated that RAS activation was involved in dyslipidemia-mediated renal injuries (14). Accordingly, after treatment with a RAS inhibitor or lipid-lowering drugs, the progression of glomerulosclerosis was attenuated (15, 16). These findings suggested the existence of synergistic effects of RAS activation and lipid disorders in renal injuries. Therefore, this study aimed to explore the potential synergistic mechanisms of these two stimuli in the development of glomerulosclerosis in HMCs.
We first investigated the effects of Ang II stimulation on intracellular cholesterol homeostasis in lipid-loaded HMCs. As demonstrated by Oil red O staining and a quantitative cholesterol assay, Ang II stimulation significantly increased intracellular lipid accumulation in HMCs. However, these effects were inhibited by telmisartan. This result is consistent with the phenomenon that the administration of Ang II in animal models induces abnormal lipid metabolism in the heart (17), liver (18), and kidney (19), as well as accelerates other target organ injuries. Additionally, a recent study revealed that the Ang II-induced profibrotic responses in human mesangial cells was partially amplified in the presence of high concentrations of free fatty acid and could be reversed by rosuvastatin (11); this result suggests that lipid loading is closely associated with Ang II-induced pathogenic effects. However, the potential mechanisms in this process were not clarified.
The low density lipoprotein receptor (LDLr), whose expression is predominantly regulated by the intracellular cholesterol pool at the transcriptional level through a negative feedback mechanism, is an important cholesterol influx receptor. A high intracellular concentration of cholesterol prevents the transport of sterol regulatory element-binding protein (SREBP) cleavage activating protein (SCAP)-SREBP-2 complexes from the endoplasmic reticulum (ER) to the Golgi and down-regulates LDLr expression (20). In this study, we demonstrated that Ang II upregulated LDLr gene transcription due to a stimulative effect on the translocation of the SCAP/SREBP2 complex from the ER to the Golgi. This result is in accordance with in vivo findings from the Saito group, who showed that the protein expression of SREBPs in the kidney of Ang II-treated rats was higher than that in controls (19). Furthermore, the studies that have investigated the role of RAS activation in lipid metabolism have mainly focused on atherosclerosis and the involved molecules, such as scavenger receptor class B type I (SR-BI) (21), lectin-like oxidized LDL scavenger receptor-1 (LOX-1) (22), and LDL receptor-related protein 1 (LRP1) (23). Because lipid homeostasis is likely to be tissue/cell specific and its regulation in kidneys is not completely understood, the potential mechanism for the disruption of the LDLr pathway in HMCs that is induced by Ang II needs to be elucidated.
In the last few years, it has become clear that mesangial cells express all components of the RAS pathway under normal physiological conditions and that the intracellular RAS may be activated in response to pathological stimuli (24). Therefore, we investigated the effects of lipid loading on the activation of the intracellular RAS. The results showed that lipid loading caused a significant upregulation of intracellular RAS components at the mRNA and protein levels in HMCs, in accordance with previous studies and supporting the opinion that cholesterol metabolites are regulators of RAS activation through certain mechanisms. Early studies demonstrated that both native and oxidized LDL increased AT1 gene expression in vascular smooth muscle cells (25) and in human coronary artery endothelial cells (26). Studies from Park et al. (27) demonstrated that LDL-induced mesangial cell proliferation was inhibited by losartan, showing that AT1 plays an important role in this process. Interestingly, in this study, both AT1 and AT2 expression in HMCs was increased after lipid loading. It is accepted that Ang II, as a principal effector of RAS, exerts a variety of responses through AT1 and AT2. The majority of Ang II-promoted responses in kidney diseases are mediated via AT1, while AT2 has been thought to counteract the effects of AT1 and to play a role in the protection of the kidney (28). Recent evidence suggests that AT2 not only opposes the actions of AT1 but also has a unique effect independent of its interaction with AT1 signaling. Wolf et al. (29) demonstrated that AT2 contributes to the recruitment of inflammatory cells through the activation of nuclear factor-κB and that the potential proinflammatory effects mediated by Ang II may not be totally eliminated by AT1 antagonists; this result suggests that the balance between AT1 and AT2 may be valuable in evaluating kidney injuries (30). In this study, we found that the ratio of AT1/AT2 increased in HMCs after lipid loading, suggesting that the balance between the two receptors was disrupted.
Mesangial cell activation is an important event in the development of glomerulosclerosis (31). Functional changes in mesangial cells, such as cell polarity, morphogenesis, migration, cell cycle progression, and cell growth or death, are common features of glomerular diseases. When injury occurs, mesangial cells rapidly synthesize ECM components, such as collagen, laminin, and fibronectin. As sclerosis progresses, there is occlusion of the capillary loops, which ultimately leads to the loss of glomerular function (2). In this study, lipid loading promoted cell growth and cell cycle progression in parallel with an increased accumulation of collagen I, α-SMA, and fibronectin synthesized by HMCs, which is an important feature of glomerulosclerosis.
This study is the first to demonstrate that Ang II induced lipid accumulation in HMCs through the disruption of the LDLr pathway and lipid loading activated the intracellular RAS to induce phenotypic changes and dysfunction in mesangial cells. The interaction between intracellular RAS activation and lipid disorders accelerated the progression of glomerulosclerosis, suggesting that the management of blood pressure and proteinuria by RAS blockers and of cholesterol by statins are not independent components of the treatment regimen.
Acknowledgements
This work was supported by Grant BK2009279 from the Natural Science Foundation of Jiangsu Province and Grants 81170792 and 81070571 from the National Natural Science Foundation of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Daskalakis N, Winn MP. Focal and segmental glomerulosclerosis. Cell Mol Life Sci. 2006;63(21):2506-2511
2. Pozzi A, Voziyan PA, Hudson BG, Zent R. Regulation of matrix synthesis, remodeling and accumulation in glomerulosclerosis. Curr Pharm Des. 2009;15(12):1318-1333
3. Mattana J, Kochlatyi S, Gibbons N. Metal-catalyzed oxidation of extracellular matrix proteins promotes human mesangial cell apoptosis and is associated with enhanced expression of Bax and caspase activation. Biochem Biophys Res Commun. 2002;292(3):652-658
4. Hua P, Feng W, Rezonzew G, Chumley P, Jaimes EA. The transcription factor ETS-1 regulates angiotensin II-stimulated fibronectin production in mesangial cells. Am J Physiol Renal Physiol. 2012;302(11):F1418-F1429
5. Keane WF. The role of lipids in renal disease: future challenges. Kidney Int Suppl. 2000;75:S27-S31
6. Singh BK, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in atherogenesis and therapy of coronary heart disease. Indian Heart J. 2001;53(4):511-518
7. Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch Intern Med. 2003;163(11):1296-1304
8. Catar RA, Muller G, Heidler J, Schmitz G, Bornstein SR, Morawietz H. Low-density lipoproteins induce the renin-angiotensin system and their receptors in human endothelial cells. Horm Metab Res. 2007;39(11):801-805
9. Wang X, Phillips MI, Mehta JL. LOX-1 and angiotensin receptors, and their interplay. Cardiovasc Drugs Ther. 2011;25(5):401-417
10. Gamble W, Vaughan M, Kruth HS, Avigan J. Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J Lipid Res. 1978;19(8):1068-1070
11. Solini A, Rossi C, Santini E, Madec S, Salvati A, Ferrannini E. Angiotensin-II and rosuvastatin influence matrix remodeling in human mesangial cells via metalloproteinase modulation. J Hypertens. 2011;29(10):1930-1939
12. Wang Z, Jiang T, Li J, Proctor G, McManaman JL, Lucia S. et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54(8):2328-2335
13. Sasatomi Y, Tada M, Uesugi N, Hisano S, Takebayashi S. Obesity associated with hypertension or hyperlipidemia accelerates renal damage. Pathobiology. 2001;69(2):113-118
14. Ni J, Ma KL, Wang CX, Liu J, Zhang Y, Lv LL. et al. Activation of renin-angiotensin system is involved in dyslipidemia-mediated renal injuries in apolipoprotein E knockout mice and HK-2 cells. Lipids Health Dis. 2013;12(1):49
15. Kostapanos MS, Liberopoulos EN, Elisaf MS. Statin pleiotropy against renal injury. J Cardiometab Syndr. 2009;4(1):E4-E9
16. Oite T. Exploring the mechanisms of renoprotection against progressive glomerulosclerosis. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(3):81-90
17. Hongo M, Ishizaka N, Furuta K, Yahagi N, Saito K, Sakurai R. et al. Administration of angiotensin II, but not catecholamines, induces accumulation of lipids in the rat heart. Eur J Pharmacol. 2009;604(1-3):87-92
18. Ishizaka N, Hongo M, Sakamoto A, Saito K, Furuta K, Koike K. Liver lipid content is reduced in rat given 7-day administration of angiotensin II. J Renin Angiotensin Aldosterone Syst. 2011;12(4):462-468
19. Saito K, Ishizaka N, Hara M, Matsuzaki G, Sata M, Mori I. et al. Lipid accumulation and transforming growth factor-beta upregulation in the kidneys of rats administered angiotensin II. Hypertension. 2005;46(5):1180-1185
20. Abrass CK. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol. 2004;24(1):46-53
21. Pilon A, Martin G, Bultel-Brienne S, Junquero D, Delhon A, Fruchart JC. et al. Regulation of the scavenger receptor BI and the LDL receptor by activators of aldosterone production, angiotensin II and PMA, in the human NCI-H295R adrenocortical cell line. Biochim Biophys Acta. 2003;1631(3):218-228
22. Hu C, Kang BY, Megyesi J, Kaushal GP, Safirstein RL, Mehta JL. Deletion of LOX-1 attenuates renal injury following angiotensin II infusion. Kidney Int. 2009;76(5):521-527
23. Sendra J, Llorente-Cortes V, Costales P, Huesca-Gomez C, Badimon L. Angiotensin II upregulates LDL receptor-related protein (LRP1) expression in the vascular wall: a new pro-atherogenic mechanism of hypertension. Cardiovasc Res. 2008;78(3):581-589
24. Kumar R, Boim MA. Diversity of pathways for intracellular angiotensin II synthesis. Curr Opin Nephrol Hypertens. 2009;18(1):33-39
25. Nickenig G, Sachinidis A, Michaelsen F, Bohm M, Seewald S, Vetter H. Upregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cells. Circulation. 1997;95(2):473-478
26. Li D, Saldeen T, Romeo F, Mehta JL. Oxidized LDL upregulates angiotensin II type 1 receptor expression in cultured human coronary artery endothelial cells: the potential role of transcription factor NF-kappaB. Circulation. 2000;102(16):1970-1976
27. Park SY, Song CY, Kim BC, Hong HK, Lee HS. Angiotensin II mediates LDL-induced superoxide generation in mesangial cells. Am J Physiol Renal Physiol. 2003;285(5):F909-F915
28. Wenzel UO, Krebs C, Benndorf R. The angiotensin II type 2 receptor in renal disease. J Renin Angiotensin Aldosterone Syst. 2010;11(1):37-41
29. Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin II activates nuclear transcription factor-kappaB through AT1 and AT2 receptors. Kidney Int. 2002;61(6):1986-1995
30. Siragy HM. AT1 and AT2 receptor in the kidney: role in health and disease. Semin Nephrol. 2004;24(2):93-100
31. Yang Y, Zhang SY, Sich M, Beziau A, van den Heuvel LP, Gubler MC. Glomerular extracellular matrix and growth factors in diffuse mesangial sclerosis. Pediatr Nephrol. 2001;16(5):429-438
Author contact
![]() Corresponding author: Bi Cheng Liu. Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, NO.87, Ding Jia Qiao Road, Nanjing City, Jiangsu Province, P.R. China, 210009. Tel: 0086 25 83262442, Fax 0086 25 83262442, Email: liubc64com
Corresponding author: Bi Cheng Liu. Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, NO.87, Ding Jia Qiao Road, Nanjing City, Jiangsu Province, P.R. China, 210009. Tel: 0086 25 83262442, Fax 0086 25 83262442, Email: liubc64com
Received 2013-5-7
Accepted 2013-8-30
Published 2013-9-18