ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2013; 10(7):812-824. doi:10.7150/ijms.5931 This issue Cite
Research Paper
Angiotensin Receptor Blockers and Statins Could Alleviate Atrial Fibrosis via Regulating Platelet-Derived Growth Factor/Rac1 /Nuclear Factor-Kappa B Axis
Dongfang Yang1,*, Jia Yuan1,*, Gan Liu1,2, Zhiyu Ling1, Haiyan Zeng3, Yunqing Chen1, ![]() , Yue Zhang1, Qiang She1, Xue Zhou4
, Yue Zhang1, Qiang She1, Xue Zhou4
1. Department of Cardiology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China;
2. Department of Cardiology, The People's Hospital of Hechuan District, Chongqing, China;
3. Department of Epidemiology and Health Statistics, The Public Health College of Huaxi of Sichuan University, Sichuan, China;
4. Department of cardiology, Children's hospital of Chongqing Medical University, Chongqing, China.
* Dongfang Yang and Jia Yuan contributed equally to this work.
Abstract
Aims: To investigate whether the administration of renin-angiotensin system (RAS) inhibitors and statins could alleviate atrial fibrosis via platelet-derived growth factor (PDGF)/Rac1 /nuclear factor-kappa B (NF-κB) axis.
Methods and Results: In human left atrium, the degree of atrial fibrosis, as well as the expression levels of PDGF, Rac1 and NF-κB increased 1.5 to 2.9 folds in patients with atrial fibrillation compared to that with sinus rhythm, (P<0.0001). There were strongly positive correlations between angiotensin II (Ang II) or procollagen type III-alpha-1 (COL3A1) with PDGF, Rac1, NF-κB, and among PDGF, Rac1 and NF-κB (all P<0.05). At 3 weeks after the transverse aorta constriction (TAC) operation in rat model and with intervention of irbesartan or/and simvastatin, the collagen volume fraction (CVF) and atrial natriuretic peptide (ANP) values respectively increased 6-folds and 3.5-folds in the TAC group compared to SHAM group (P<0.0001), but these levels decreased by 16% to 63% with following drug intervention (all P<0.0001), the combined treatment was the lowest. Accordingly, the expression levels of PDGF (3-folds), Rac1 (1.6-folds), NF-κB (7-folds) and AngII (12-folds) significantly increased in the TAC group compared to the SHAM group, and these levels were also reduced by 25% to 64% with following drug intervention. The highest reduction could be seen after treatment with irbesartan and simvastatin in combination (all P<0.001).There were strongly positive correlations between AngII or CVF with PDGF, Rac1, NF-κB, and among PDGF, Rac1 and NF-κB (all P<0.05).
Conclusions: Irbesartan or/and simvastatin can improve atrial fibrosis by regulating PDGF/Rac1/NF-κB axis.
Keywords: atrial fibrosis, platelet-derived growth factor (PDGF), Rac1, nuclear factor-kappa B (NF-κB), Irbesartan, Simvastatin.
Introduction
Atrial fibrosis is one of the most fundamental mechanisms involved in the physiopathology of cardiovascular disease, which includes atrial fibrillation (AF)[1,2] and heart failure (HF)[3]. The specific mechanism responsible for atrial fibrosis remains unclear. Many recent studies in both animal models and humans have revealed that renin-angiotensin system (RAS) activation is one of the most vital mechanisms involved in atrial fibrosis[1,4]. Angiotensin II (Ang II) activation initially activates the RAS, which is involved in the pathogenesis of atrial fibrosis via the activation of downstream signal transduction pathways, such as mitogen-activated protein kinase (MAPK) pathways[1,4,21], Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathways[14] and transforming growth factor-β1 (TGF-β1) pathways[5,6].
In addition, other studies demonstrated that AngII activated platelet-derived growth factor-A (PDGF-A), a novel contributor to cardiac remodelling, to result in atrial fibrosis in pressure-overloaded mouse model [13]. Burstein et al [7] found that PDGF had a tendency to promote atrial fibrosis in dogs. The expression and activity levels of Rac1, a small GTP-binding protein, and nuclear factor-kappa B (NF-κB), a transcription factor, increased in fibrillating human atrial tissues[18,19]. In addition, Rac1 may activate NF-κB[8] and STAT3[1,4,12], and both of these signalling pathways can be activated by AngII to play important roles in atrial fibrosis[9]. Statins, which are 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, can reduce Rac1 activity and thereby contribute to the improvement of atrial fibrosis and the prevention of AF, which has been verified in many animal and clinical studies[6,10-11]. Furthermore, angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs), which were initially used to treat hypertension, have now been used to treat atrial fibrosis via inhibiting Angiotensin[15-17].
Sulciner et al [8] reported that Rac1 protein leaded to activation of NF-κB in HeLa cell. Whether Rac1 protein activating NF-κB involves in mechanism of atrial fibrosis is unclear. PDGF and Rac1 could be activated by Ang II, but the relationship between PDGF and Rac1 is also unclear now. Thus, we hypothesized that PDGF, Rac1 and NF-κB maybe formed a signal transduction pathway axis, which could be inhibited by RAS inhibitors or/and statins, thereby participating in the mechanism of atrial fibrosis.
By now, all of previous researches about PDGF inducing atrial fibrosis were based on animal experiments, rather than on human. The aim of the present study was to assess the relationship among PDGF, Rac1 and NF-κB in fibrotic atrium induced by a rat model of transverse aorta constriction (TAC) and human fibrillating atrium, and to evaluate the beneficial effects of angiotensin receptor blockers (irbesartan) or/and Rac1 inhibitors (simvastatin) on atrial fibrosis via PDGF/Rac1/NF-κB axis.
Methods
Collection of human atrial tissues
The patients included in this study ranged from 18 to 65 years of age and suffered with valvular heart disease (VHD) required valve replacement at the Zhong Shan Hospital of Chongqing from November 2010 to December 2011. 12 Patients with AF and 12 patients with sinus rhythm (SR) were included. The protocol was conducted under the Helsinki Declaration and was approved by the ethics committees of the Second Hospital of Chongqing Medical University. Prior to surgery, informed consent was acquired from each patient included in this study. Left atrial tissue from each patient was acquired from the cannulation position following the termination of extracorporeal circulation, and the tissues were immediately stored in liquid nitrogen for subsequent experiments.
Transverse aorta constriction (TAC) animal model
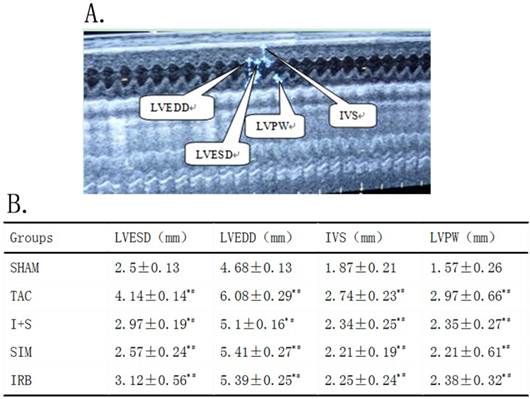
The TAC operation was performed on adult Wistar rats weighing approximately 220 g, and the rats were then divided equally into 5 groups (n=12 for each group). The SHAM group was maintained on a standard diet and was subjected to a thoracotomy without the TAC operation. The TAC group was maintained on a standard diet and was subjected to the TAC operation. The irbesartan (IRB), simvastatin (SIM) and IRB+SIM (I+S) groups were treated with irbesartan (Sanofi, France, 20 mg/kg/d), simvastatin (MSD, USA, 5 mg/kg/d), a combination of irbesartan (20 mg/kg/d) and simvastatin (5 mg/kg/d), which was injected into the stomach through a nasogastric tube on next day after TAC operation. The precise TAC operation method was conducted following by Chien-hui Liao[13]. Two weeks after the drugs being administered, the left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic diameter (LVESD), interventricular septum (IVS) and left ventricular posterior wall (LVPW) were detected using a GE Vingemed ultrasound (USA) and a 13 MHZ frequency probe. At three weeks after the drugs being administered, the atrial tissues were collected and divided into two pieces, which were stored separately in liquid nitrogen or 4% paraformaldehyde. Experimental animal handing was performed according to the rules of the American Heart Association (AHA).
Trinitrophenol sirius red dyes
Following fixation in 4% paraformaldehyde for 24 hrs, the atrial tissues were subjected to routine dehydration, paraffin embedding, dewaxing (to hydrate) and dying for 5-10 min in Lapis lazuli blue liquid. The samples were then washed 3 times and dyed for 15-30 min in trinitrophenol Sirius dyeing liquid prior to re-dehydration. Finally, xylene was used to clear the tissues, and they were embedded in neutral balsam solid. Collagen was detected from different fields of individual sections using polarised light microscopy (Olympus BX51TR, Japan), and the collagen volume fraction (CVF) of each image was analysed using Image-Pro Plus 6.0 software (Media Cybernetics, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from tissues with Tripure Reagent(Roche, Switzerland), chloroform and isopropanol, then complementary deoxyribonucleic acid (cDNA) was synthesized by Reverse Transcription Kit (Promega, USA). RT-PCR amplification reactions were implemented by PCR amplifier (BioRad-MyClycler M367960, USA), and then all products were conducted gel electrophoresis using Bio-Rad PowerPac Basic electrophoresis apparatus (USA) and analysed pictures by Quantity One software version 4.6.2 (Bio-Rad, USA). Base sequences of primers of target gene had exactly exhibited in Table 1 (Sangon, China).
Enzyme-linked immunosorbent assay (ELISA)
Total protein was extracted from tissues and the mixed solution was centrifuged at 1000 rpm for 10 min to eliminate granules and polymers. Then, the following steps were conducted by the manufacturer's recommendations (DAA00B R&D company, USA). Finally, the OD (optical density) of each well was immediately assayed at 450 nm using a micro-plate reader (Thermo Fisher 1510, USA).
Radioimmunoassay
Concentration of Ang II was tested by Radioimmunoassay, which were conducted by the manufacturer's recommendations (Western company, China). The cpm values of the deposit were tested by microplate reader.
Rac1 GST-p21-activated kinase (GST-PAK) pull-down assay
The activated Rac1 was detected by GST-PAK pull-down assay, which was conducted by the manufacturer' recommendations (Upstate Biotechnology, USA). Activated Rac1 was detected by chemiluminescent detection system (Bio-Rad, California, USA) and All immunoblotting results were exposed to ultraviolet ray and analyzed by quantity one 4.6.2 software (Bio-Rad, California, USA).
Primers of target gene.
| Target gene | Primer | Base sequence | |
|---|---|---|---|
| COL1A1 | sense | 5'-GTGTTGCTGAAAGACTACCTCGT-3' | |
| antisense | 5'-GGTACAGAGTCTTTTGCTTCCTC-3' | ||
| ANP | sense | 5'-CTCAGAGAGATGGAGGTGCTCTC-3' | |
| Rat | antisense | 5'-CTCAGAGAGATGGAGGTGCTCTC-3' | |
| PDGFb | sense | 5'-CTCAGAGAGATGGAGGTGCTCTC-3' | |
| antisense | 5'-GCCCAGAGGAGTTCATGTCTTAT-3' | ||
| Beta-actin | sense | 5'-GTACAACCTTCTTGCAGCTCCT-3' | |
| antisense | 5'-ACAGGATTCCATACCCAGGAA-3' | ||
| COL3A1 | sense | 5'-GTACTCCTGGTCTGCAAGGAATG-3' | |
| antisense | 5'-GGGTCCTGGGTTACCATTACTAC-3' | ||
| COL1A1 | sense | 5'-CTCAAGATGTGCCACTCTGACT-3' | |
| antisense | 5'-GAGGGAGTTTACAGGAAGCAGAC-3' | ||
| Human | TIMP1 | sense | 5'-GCGTGGACATTTATCCTCTAGC-3' |
| antisense | 5'-AGCCAACAGTGTAGGTCTTGGT-3' | ||
| PDGFb | sense | 5'-AATAGGAGGGAGACTGTGGTAGG-3' | |
| antisense | 5'-AGAAGGATCAAAGATGGGGAAGC-3' | ||
| Beta-actin | sense | 5'-CTTCTACAATGAGCTGCGTGTG-3' | |
| antisense | 5'-AGTCATAGTCCGCCTAGAAGC-3' |
Note: COL1A1, procollagen type I-α-1; ANP, atrial natriuretic peptide; PDGFb, platelet-derived growth factor-b; COL3A1, procollagen type III-α-1; TIMP-1, tissue inhibitor metalloproteinase-1.
Electrophoretic mobility shift assays (EMSAs)
The activity of NF-κB was tested by EMSAs, which was performed according to the manufacturer's instructions for the LightShift Chemiluminescent EMSA kit (Pierce 20148, USA). The NF-κB consensus sequence (5'-AGT GGG GAC TTT CCC AGGC-3' and 3'-TCA ACT CCC CTG AAA GGG TCC-5') was biotin-labelled and purified (Beyotime GS056B). Polyacrylamide gel electrophoresis was used to separate the target protein-DNA compounds, and then the target compounds were detected via chemiluminescence.
Measurement of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity using a colorimetric quantitative detection assay
Measurement of NADPH was followed by the manufacturer's instructions provided with the GENMED kit (GMS50096.2, USA). NADPH oxidase activity was evaluated using a colorimetric assay and a spectrophotometer.
Statistical analyses
All of the data are presented as the means±SD and were analysed using the SAS statistical analysis package (venison 8.0, SAS company, USA). The basic patient data were analysed with an unpaired Student's t-test. The data from the model animals were analysed with a one-way ANOVA, and two groups were compared using an unpaired Student's t-test. Correlation analysis were used to estimate the relation between atrial expression of Ang II, CVF, COL3A1, ANP, COL1A1, NADPH, TIMP1 with the expression levels of PDGFb, PDGF-BB, Rac1, NF-κB in left atrial tissue. A probability value of <0.05 was considered statistically significant.
Results
Atrial fibrosis was successfully induced by atrial pressure overload in a TAC rat model, and fibrosis was partially relieved following treatment with irbesartan or/and simvastatin
To create an atrial fibrosis model following cardiac hypertrophy, we firstly induced transverse aorta constriction (TAC) in a rat model and then intervened with irbesartan, simvastatin or a combination of irbesartan and simvastatin (n=12 in each group). At 2 weeks after the procedure, the TAC group and the drug intervention groups demonstrated a significant increase in LVESD, LVEDD, IVS and LVPW in comparison to the SHAM group (P<0.0001) (Fig.1A). These data indicated that the animal TAC model was successfully established. Moreover, the LVSD, LVDD, IVS and PW values of the SIM, IRB and I+S groups were significantly decreased 11% to 38% when compared to the TAC group (P<0.05) (Fig.1B). However, there was no differences in LVSD, LVDD, IVS and PW values among SIM, IRB and IRB+SIM groups (P>0.05) (Fig.1B).
(A) Representative echocardiograph photo indicated how to measure LVESD, LVEDD, IVS, LVPW in rats. (B) when compared with SHAM group, there was * p<0.05, but compared with TAC group, there was # p<0.05. n=12 for each group. LVESD, left ventricular systolic diameter; LVEDD, left ventricular end-diastolic dimension; IVS, interventricular septum; LVPW, left ventricular posterior wall.
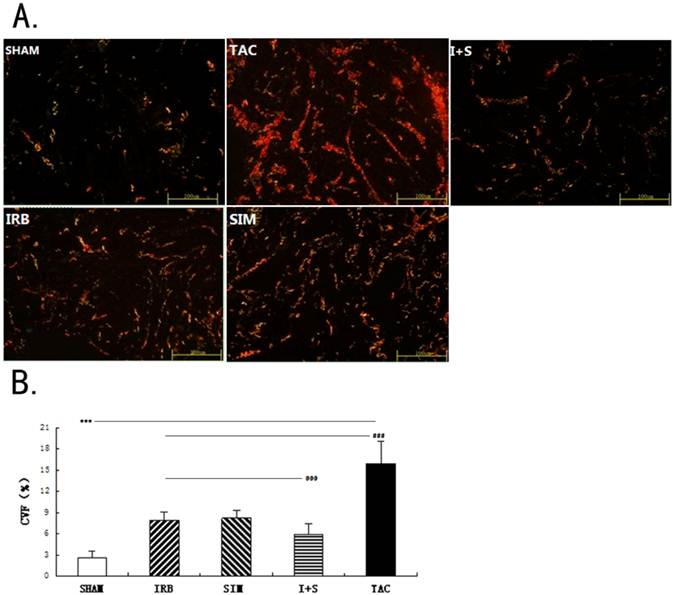
At three weeks after the TAC operation, changes in atrial fibrosis were detected via Sirius red staining. The CVF included the red, green and yellow areas (Fig.2A). The CVF values of the TAC group (15.92%) was the highest in all experiment groups, following I+S group, SIM group, IRB group, SHAM group (5.97%, 8.23%, 7.92% and 2.63%, respectively. n=12 in each group) (P<0.0001). Through drug intervention, the expression levels of CVF was effectively decreased 48% to 63% compared to the TAC group, the I+S group was the most reduction (P<0.0001) (Fig.2B).
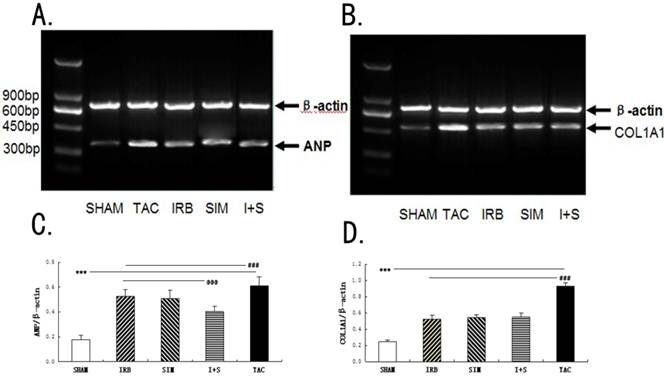
Similarly, as shown in Fig.3A-3B, by RT-PCR analysis, the COL1A1(0.93±0.04) and ANP(0.61±0.07) values of the TAC group was the highest in all experiment groups. In drug intervention groups, the COL1A1 and ANP were reduced 16% to 44% compared to TAC group, and the most reduction was seen in the I+S group (Fig.3C-3D).
Expression of procollagen type I-alpha-1 (COL1A1) and procollagen type III-alpha-1 (COL3A1) in left atral tissue of rats. (A).Representative micrographs of Trinitrophenol Sirius red dyeing exhibiting the interstitial collagen including COL1A1 (stained as red), COL3A1 (stained as green) and the merging of COL1A1 with COL3A1 (stained as yellow). Magnification ×400. Scale bar=100μm. (B). The collagen volume fraction (CVF) included the red, green and yellow areas. The CVF is expressed by histogram. n=12 for each group. *** p<0.001 versus other four groups, respectively; ### p<0.001 versus IRB or/and SIM groups, respectively;@@@ p<0.001 versus IRB or SIM groups, respectively.
Expression of atrial natriuretic peptide (ANP) and procollagen type I-alpha-1 (COL1A1) in left atrial tissue of rats. (A, B) Expression of ANP and COL1A1 in RAAs analyzed by RT-PCR. (A) Representative bands of ANP. (B) Representative bands of COL1A1. (C) Quantitative expression of ANP by histogram. (D) Quantitative expression of COL1A1 by histogram. β-actin is an internal standard. Values are mean ± SEM and are expressed relative to an average of SHAM group. n=12 for each group. *** p<0.001 versus other four groups, respectively; ### p<0.001 versus IRB or/and SIM groups, respectively; @@@ p<0.001 versus IRB or SIM groups, respectively. β-actin: beta-actin.
Clinical characteristics of patients.
| AF (n=12) | SR (n=12) | P value | |
|---|---|---|---|
| sex, M/F(n) | 3\9 | 4\8 | 1 |
| Age(years) | 47.6±9.9 | 47.7±9.5 | 0.972 |
| NYHA class I/II/III/IV | 0\1\8\3 | 0\1\9\2 | 0.8786 |
| ECH | |||
| LVDd(mm) | 52±11.7 | 51.8±12 | 0.971 |
| EF(%)* | 52.3±4.6 | 58.4±6.5 | 0.015 |
| LAD(mm)** | 54.3±11 | 39.5±4.9 | <0.001 |
| RAD(mm) | 37.9±15.3 | 39.2±9.5 | 0.816 |
| RVD(mm) | 25±8.5 | 21.4±3.9 | 0.209 |
| LVPWT(mm) | 9.8±2.1 | 9.6±2 | 0.82 |
| IVSd(mm) | 9.5±1.2 | 10±1.8 | 0.442 |
| Cause of valve disease(n) | |||
| Rheumatic | 8 | 6 | 0.4076 |
| Preoperative drugs(n) | |||
| ACEI/ARB | 6 | 5 | 0.682 |
| Calcium-channel blocker | 2 | 2 | 1 |
| Beta-blocker** | 10 | 3 | 0.0041 |
| Diuretic | 8 | 9 | 1 |
| Digitalis | 10 | 9 | 1 |
| Statin | 8 | 5 | 0.2191 |
| Anticoagulant/antiplatelet | 11 | 8 | 0.3168 |
Values are mean ± SD or number of patients. ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; NYHA, New York Heart Association; LVDd, left ventricular enddiastolic diameter; EF, ejection fraction; LAD, left atrial diameter; RAD, right atrial diameter; LVPWT, left ventricular posterior wall thick ness; IVSd, interventricular septum diameter. * p<0.05 or ** p<0.01 between groups, reference group is the SR group.
The concentration of Ang II and the expression levels of PDGF, activated-Rac1 and NF-κB were increased following atrial fibrosis induction in TAC rat model
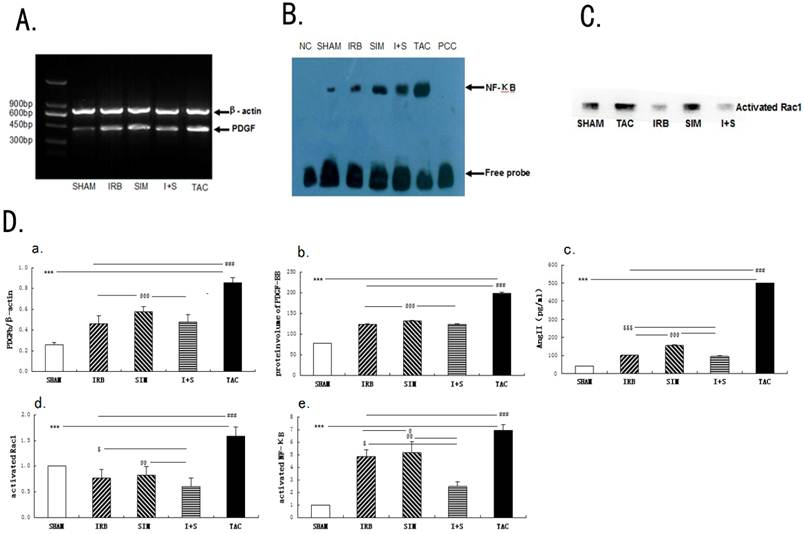
After the rat TAC model being successfully established for 3 weeks, at which point atrial fibrosis had been induced. The Ang II concentration increased 12 folds in the TAC group compared to the SHAM group. After administration of irbesartan or/and simvastatin, the concentration of Ang II notably decreased 69% to 81% compared to TAC group. This decrease was more pronounced in the I+S group. (All P<0.001, n=12 in each group) (Fig.4D).
The ELISA analysis revealed that the expression of PDGF-BB protein significantly increased 2.6 folds, 1.6-folds,1.6-folds,1.7 folds in the TAC, I+S, IRB and SIM groups in comparison to the SHAM group(all P<0.0001, n=12 in each group) (Fig. 4D). These similar results were verified by using RT-PCR analysis for PDGFb mRNA expression levels (all P<0.0001) (Fig.4A and 4D).
Based on the PAK-PBD pull-down assay, the expression level of activated Rac1 increased about 1.6 folds in the TAC group compared to SHAM group (P<0.001) (Fig.4C-4D). Following treatment with irbesartan or/and simvastatin, the expression level of activated Rac1 decreased significantly about from 48% to 63% in comparison to TAC group (P<0.001) (Fig.4D). Moreover, the I+S group (0.6±0.17) displayed the lowest activated Rac1 expression level (Fig.4D).
Based on the EMSA results, the expression level of NF-κB increased about 7 folds in the TAC group compared to SHAM group (P<0.001) (Fig.4B). Following treatment with irbesartan or/and simvastatin, the NF-κB expression level decreased significantly about from 25% to 64% in comparison to that measured in the TAC group (P<0.001) (Fig.4D). The reduction reached peak in the I+S group (Fig.4D).
Human atrial samples
The clinical characteristics of the patients studied are presented in Table 2. The AF patients demonstrated a lower ejection fraction (P=0.015), higher left atrial diameter (LAD) values (P<0.001) and greater preoperative use of beta-blockers (all P=0.0041) than the SR patients. No significant differences were observed in other parameters (P>0.05).
Expression levels of NADPH, COL1A1, COL3A1 and tissue inhibitor of metalloproteinase-1 (TIMP-1) in human Left atrial samples
Based on RT-PCR analysis, there were differences in the mRNA levels of COL1A1(1.05±0.06 v 0.6±0.05), COL3A1(1.12±0.09 v 0.8±0.1) and TIMP-1 (0.8±0.1 v 0.6±0.09) between the AF patients and SR patients (P<0.001,Fig.5A-5D). And the activity of NADPH in the AF patients (as tested by colorimetry) increased 12 folds compared to the SR patients (P=0.0002) (Fig.6D).
Activation of the PDGF/Rac1/ NF-κB signalling axis in human Left atrial samples
The expression levels of PDGFb(1.05±0.1 v 0.52±0.06, by RT-PCR), PDGF-BB (8.77±0.36 v 3.49±0.15, by ELISA), Rac1 (2.76±1.07 v 1.5±0.46, by the GST-PAK pull-down assay) and NF-κB (5.4±2.03 v 1.84±0.98,by EMSA) were higher in AF patients than that in SR patients (P<0.01,Fig.6A-6D).
Expression of platelet-derived growth factor (PDGF), Rac1 and nuclear factor-kappa B NF-κB in left atrial of rats. (A-C) Expression of PDGF, Rac1 and NF-κB in RAAs analyzed by ELISA, RT-PCR, GST-PAK pull down assay and EMSAs, respectively. (A)Representative bands of PDGF, n=12 for each group (B) Representative bands of NF-κB, n=12 for each group (C) Representative bands of Rac1, n=12 for each group. (D) Quantitative expression of AngII (Dc), protein and mRNA volume of PDGF (Da and Db), NF-κB(De) and activated Rac1(Dd) in left atrial samples. β-actin is an internal standard. Values are mean ± SD. *** p<0.001 versus other four groups, respectively; ### p<0.001 versus IRB or/and SIM groups, respectively; @@@ p<0.001 versus IRB or SIM groups, respectively; @@ p<0.01 versus I+S group; @ p<0.05 versus IRB group; $$$ p<0.001 versus I+S group; $ p<0.05 versus I+S group, respectively. PCC, probe cold competition. NC, negative control.
Expression of procollagen type I-alpha-1 (COL1A1), procollagen type III-alpha-1 (COL3A1) and tissue inhibitor metalloproteinase-1 (TIMP-1) in human RAAs. (A-C) Expression of COL1A1, COL3A1 and TIMP-1 in RAAs analyzed by RT-PCR. (A) Representative bands of COL1A1. (B) Representative bands of COL3A1. (C) Representative bands of TIMP-1. (D) Quantitative expression of COL1A1, COL3A1 and TIMP-1 in RAAs. β-actin is an internal standard. Values are mean ± SD. n=12 for each group. *** p<0.001 between groups, reference group is the SR group.
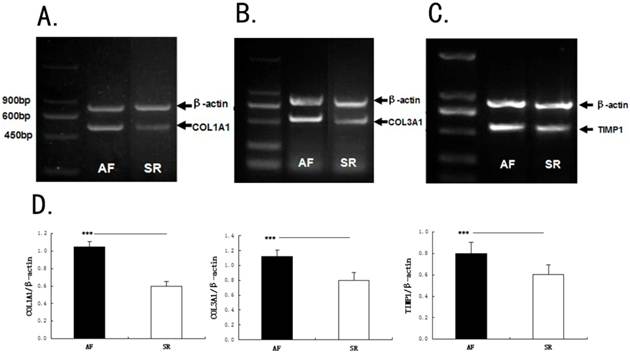
Expression of platelet-derived growth factor (PDGF), Rac1 and nuclear factor-kappa B NF-κB in human RAAs. (A-C) Expression of NADPH, PDGF, Rac1 and NF-κB in human RAAs analyzed by Colorimetric quantitative detection assay, ELISA, RT-PCR, GST-PAK pull down assay and EMSAs, respectively. (A) Representative bands of PDGF; (B)Representative bands of NF-κB; (C) Representative bands of Rac1; (D) Quantitative expression of PDGF, activated Rac1, NF-κB, NADPH and AngII in RAAs of cardiac disease patients with AF matched with patients with SR. N=12 for each groupβ-actin is an internal standard. Values are mean ± SEM. ** p<0.01 between groups; *** p<0.001 between groups, reference group is the SR group.
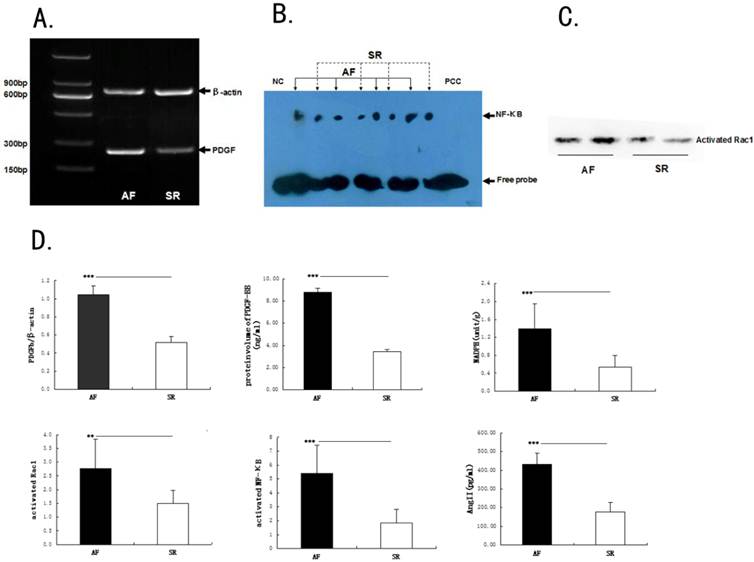
Correlations between Ang II, CVF, COL3A1, ANP, COL1A1, NADPH, TIMP1 with the expression levels of PDGFb, PDGF-BB, Rac1, NF-κB axis in TAC rat models and left human tissue
Based on the correlations analysis, there were highly positive correlations between atrial fibrosis parameters (Ang II, ANP, CVF, COL3A1, COL1A1, NADPH, TIMP-1) with PDGFb, PDGF-BB, activated Rac1, activated NF-κB in TAC rat models or human, respectively (FIG.7A-7B,8A-8B). Simultaneously, strongly positive correlations among PDGFb, PDGF-BB, activated Rac1, activated NF-κB were found in TAC rat models or human, respectively (FIG.7C-7D, FIG.8C-8D). The precise correlation coefficient had been showed in Table 3, part data were not shown in figure. (All P<0.05).
Correlations between AngII or CVF with PDGFb, PDGF-BB, activated Rac1, activated NF-κB in rat models, respectively. (A) Linear 1 is represented as the correlations between PDGFb or activated Rac1 with AngII, and Linear 2 is represented as the correlations between PDGF-BB or activated NF-κB with AngII; (B) Linear 1 is represented as the correlations between PDGFb or activated Rac1 with CVF, and Linear 2 is represented as the correlations between PDGF-BB or activated NF-κB with CVF; (C) Linear 1 is represented as the correlations between activated Rac1 or activated NF-κB with PDGFb, and Linear 2 is represented as the correlations between activated Rac1 or activated NF-κB with PDGF-BB; (D) Linear 2 is represented as the correlation between activated Rac1 with activated NF-κB.
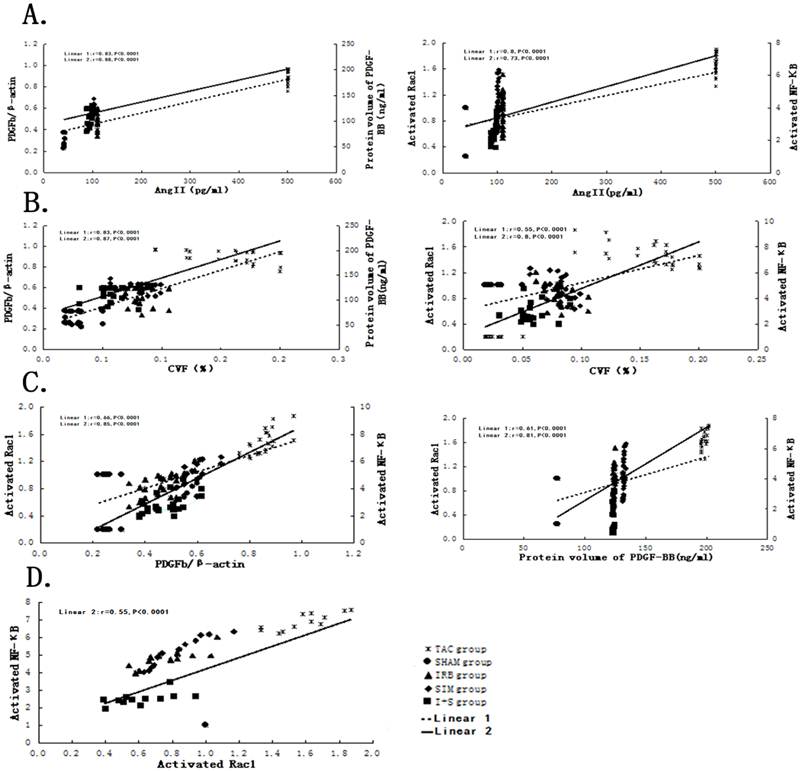
Correlations between AngII or COL3A1 with PDGFb, PDGF-BB, activated Rac1, activated NF-κB in human tissue, respectively. (A) Linear 1 is represented as the correlations between PDGFb or activated Rac1 with AngII, and Linear 2 is represented as the correlations between PDGF-BB or activated NF-κB with AngII; (B) Linear 1 is represented as the correlations between PDGFb or activated Rac1 with COL3A1, and Linear 2 is represented as the correlations between PDGF-BB or activated NF-κB with COL3A1; (C) Linear 1 is represented as the correlations between activated Rac1 or activated NF-κB with PDGFb, and Linear 2 is represented as the correlations between activated Rac1 or activated NF-κB with PDGF-BB; (D) Linear 2 is represented as the correlation between activated Rac1 with activated NF-κB.
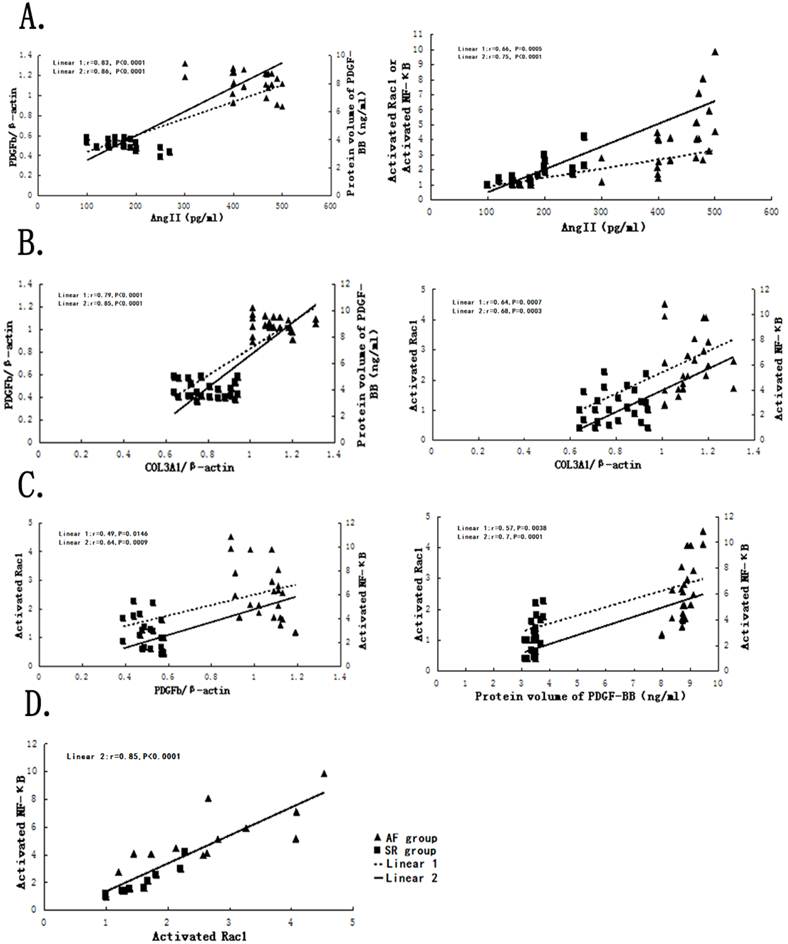
Correlations between Ang II, COL3A1, CVF, ANP, COL1A1, NADPH, TIMP1 and the expression levels of PDGFb, PDGF-BB, Rac1, NF-κB axis in TAC rat models and human tissues.
| Correlation Coefficient | ||||
|---|---|---|---|---|
| PDGF mRNA | PDGF protein | Activated Rac1 | Activated NF-κB | |
| Rat | ||||
| Ang II | 0.83*** | 0.88*** | 0.8*** | 0.73*** |
| CVF | 0.83*** | 0.87*** | 0.55*** | 0.8*** |
| Activated Rac1 | 0.66*** | 0.61*** | --- | 0.55*** |
| Activated NF-κB | 0.85*** | 0.81*** | 0.55*** | --- |
| ANP | 0.78*** | 0.82*** | 0.59** | 0.75*** |
| COL1A1 | 0.79*** | 0.81*** | 0.57*** | 0.74*** |
| Human | ||||
| Ang II | 0.83*** | 0.87*** | 0.66*** | 0.75*** |
| COL3A1 | 0.79*** | 0.85*** | 0.64*** | 0.68*** |
| Activated Rac1 | 0.49* | 0.57** | --- | 0.87*** |
| Activated NF-κB | 0.64*** | 0.69*** | 0.87*** | --- |
| COL1A1 | 0.85*** | 0.87*** | 0.73*** | 0.8*** |
| NADPH | 0.82*** | 0.74*** | 0.71*** | 0.69*** |
| TIMP-1 | 0.84*** | 0.76*** | 0.64*** | 0.59*** |
PDGF, platelet-derived growth factor; NF-κB, nuclear factor-kappa B; COL3A1, procollagen type III-alpha-1; COL1A1, procollagen type I-alpha-1; CVF, collagen volume fraction; Ang II, angiotensin II; ANP, Atrial Natriuretic Peptide; NADPH, nicotinamide adenine dinucleotide Phosphate; TIMP-1, Tissue Inhibitor Of Metallopoteinase -1. * p<0.05, ** p<0.01,*** p<0.001.
Discussion
Main findings
Based on data derived from human and TAC rat model, more serious atrial fibrosis were found in patients with AF or TAC rat model, and more serious atrial fibrosis could be improved by Irbesartan or/and simvastatin, and the most effect could be seen after treatment with irbesartan and simvastatin in combination. Furthermore, among PDGF, Rac1, NF-κB had strongly positive correlations, and may consist of an axis in signal transduction pathway inducing atrial fibrosis. These data strongly indicated that angiotensin inhibitors and statins can inhibit PDGF/Rac1/NF-κB axis and thereby can improve atrial fibrosis. Being different from previous studies, we found that PDGF involved in atrial fibrosis in human and animal models rather than animal model only, and that PDGF, Rac1, NF-κB may consist of an axis in signal transduction pathway inducing atrial fibrosis, and that administration of angiotensin inhibitors and statins with combination showed more effective than alone. These results provide that specific explanations for precise signal transduction pathways involving with atrial fibrosis and some certain support of theoretical foundation for the clinical use of angiotensin receptor blockers or/and statins to improve atrial fibrosis.
Roles of Ang II, PDGF, Rac1 and NF-κB in atrial fibrosis
At present, the precise mechanism of atrial fibrosis has remained unclear. However, Ang II acts as an initial factor to activate some signal transduction pathways, including MAPKs, JAK/STAT3, TGF-β1 and so on.
Recently, several studies [7, 22] indicated that PDGF may represent a novel potential mediator of atrial fibrosis and act as a downstream factor of Ang II. In an vitro experiment, Simm and Diez et al.[22] demonstrated that Ang II could stimulate cell proliferation in adult cardiac fibroblasts, simultaneously, and this cell proliferation could be completely inhibited by an anti-PDGF-AA antibody. In addition, a study by Burnstein et al.[7] confirmed that PDGF played a highly selective role in promoting atrial fibrosis. Some literatures revealed that activated Rac1 and NF-κB could induce atrial fibrosis and lead to atrial fibrillation in mice and humans [12, 18, 19]. Previous studies about PDGF inducing atrial fibrosis were based on vivo and vitro basic experiment. Being different from previous studies, it was firstly reported in human and TAC rat model that PDGF, Rac1 and NF-κB involved in atrial fibrosis.
Relationships among PDGF, Rac1 and NF-κB
Sulciner [8] demonstrated in HeLa cells that Rac1 protein leaded to the activation of NF-κB and regulated the ROS production. But this research did not indicate that Rac1 leading to activation of NF-κB involved in atrial fibrosis. Yoshiharu Higuchi[23] further demonstrated that Rac1 induced cardiac myocyte hypertrophy by activating NF-κB in transgenic rats. In present study, we found that Rac1 positively related to NF-κB in TAC rats and human. It could be deduced that Rac1 induced atrial fibrosis by leading to activation of NF-κB. There were rare studies to investigate the relationship of PDGF and Rac1. Therefore, the relationship between PDGF and Rac1 was also unclear. PDGF was positive correlation with Rac1 in our study of TAC rats and human (r≈0.5-0.6, P<0.05, Table 3). Moreover, Activation of PDGF and Rac1 by Ang II share common signal transduction pathways[1,4]. Thus, PDGF, Rac1 and NF-κB had tightly relationship, and may constitute a signal pathway axis, which contribute to atrial fibrosis. To our best knowledge, this axis was firstly reported to promote atrial fibrosis.
Drug intervention in atrial fibrosis
Many basic experiments and clinical studies have demonstrated that statins and angiotensin inhibitors, including ACEIs and ARBs, can improve atrial fibrosis by intervening the Ang II activation and Rac1 activation [15, 17, 20-21, 24-27]. But the precise mechanism remains unclear. In our clinical and basic experiments, AF patients and TAC rat models demonstrated an increased Ang II concentration and more serious atrial fibrosis (represented by CVF, COL3A1, COL1A1, TIMP-1), accompanying with the PDGF, Rac1, NF-κB increasing. In TAC rat model with irbesartan or/and simvastatin intervention after 3 weeks, the expression levels of Ang II, PDGF, Rac1, NF-κB, and the degree of atrium fibrosis were lighter than TAC group which indicated that irbesartan or/and simvastatin contributed to preventing from progression of atrial fibrosis, through regulating expression of PDGF, Rac1 and NF-κB. In addition, we found that the expression levels of Ang II and PDGF more reduced in group with irbesartan treatment than with simvastatin treatment and the expression of activated Rac1 was reduced in all drug intervention group, which meant that irbesartan inhibited the activation of Ang II, thereby attenuating PDGF, Rac1 and NF-κB expression, but simvastatin, which mainly attenuated the activation of Rac1 and NF-κB, was not direct useful on Ang II and PDGF. And the effect of irbesartan and simvastatin in combination was superior to the effect of irbesartan or simvastatin alone, which could be explained by that irbesartan and simvastatin separately intervened different locus of AngII/PDGF/Rac1/NF-κB axis. This result was similar to Chia-Ti Tsai[14]. But there was difference between our study and Tsai's study in intervention with different signal transduction pathways. The pathway of Tsai's study was Rac1/STAT3 axis. Another difference was that our study was based on human and animal model, but Tsai's study was based on in vivo and in vitro basic experiment.
Limitations
Certain limitations were presented in the current study. First, due to the difficulty of collecting human atrial tissue samples, we gathered appropriate tissue samples from only 24 patients, which resulted in a small sample size. Due to the restricted volume of each atrial tissue obtained, some protein expression levels were only tested by a single method. Second, because different doctors performed the operations, the samples may have been obtained from different left atrial areas. Thus, there may have been discrepancies regarding the protein expression levels between patients. Third, the interactions between Ang II, PDGF, Rac1 and NF-κB were not confirmed, and following cell culture experiments have been planned to confirm these interactions.
Conclusions
From this study on human and rat model, it demonstrated that treatment with irbesartan or/and simvastatin may improve atrial fibrosis by regulating the expression of Ang II/PDGF/Rac1/NF-κB axis, the administration of irbesartan and simvastatin in combination have more effective.
Acknowledgements
This work was jointly supported by grants from the National Natural Science Foundation of China (No. 81070140 and NO.30971213) and the project of the Chongqing Health Administration (No.2008-2-45).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802-809
2. Corradi D, Callegari S, Maestri R, Benussi S, Alfieri O. Structural remodeling in atrial fibril- ation. Nat Clin Pract Cardiovasc Med. 2008;5:782-796
3. Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodelin during atrial fibrillation. Cardiovasc Res. 2002;54:230-246
4. Nattel S, Burstein B, Dobrev D. Atrial Remodeling and Atrial Fibrillation: Mechanisms and Implications. Circ Arrhythm Electrophysiol. 2008;1:62-73
5. Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423- 432
6. Verheule S, Sato T, Everett TIV. et al. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by over-expression of TGF-beta1. Circ Res. 2004;94:1458-1465
7. Burstein B, Libby E, Calderone A, Nattel S. Differential Behaviors of Atrial Versus Ventricular Fibroblasts: A Potential Rolefor Platelet-Derived Growth Factor in Atrial-Ventricular Remodeling Differences. Circulation. 2008;117:1630- 41
8. Sulciner DJ, Irani K Yu ZX, Ferrans VJ Goldschmidt-Clermont P, Finkel T. Rac1 regulates a cytokine-stimulated redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol. 1996;16:7115-7121
9. Adam O, Frost G, Custodis F. et al. Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol. 2007;50:359-367
10. Tveit A, Grundtvig M, Gundersen T. et al. Analysis of pravastatin to prevent recurrence of atrial fibrillation after electrical cardioversion. Am J Cardiol. 2004;93:780-782
11. Young-Xu Y, Jabbour S, Goldberg R. et al. Usefulness of statin drugs in protecting against atrial fibrillation in patients with coronary artery disease. Am J Cardiol. 2003;92:1379-1383
12. Gao G, Dudley SC Jr. Redox regulation, NF-kappa B, and atrial fibrillation. Antioxid Redox Signal. 2009;11:2265-77
13. Chien-hui Liao, Hiroshi Akazawa, Masaji Tamagawa, Kaoru Ito. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J Clin Invest. 2010;120:242-253
14. Tsai CT, Lai LP, Kuo KT. et al. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: implication for the therapeutic effect of statin in atrial structural remodeling. Circulation. 2008;117:344-55
15. Ehrlich JR, Hohnloser SH, Nattel S. Role of angiotensin system and effects of its inhibition in atrial fibrillation: clinical and experimental evidence. Eur Heart J. 2006;27:512-518
16. Conen D, Tedrow UB, Koplan BA, Glynn RJ, Buring JE, Albert CM. Influence of systolic and diastolic blood pressure on the risk of incident atrial fibrillation in women. Circulation. 2009;119:2146-2152
17. Yin YH, Dalal D, Liu ZC. et al. Prospective randomized study comparing amiodarone vs. amiodarone plus losartan vs. amiodarone plus perindopril for the prevention of atrial fibrillation recurrence in patients with lone paroxysmal atrial fibrillation. Eur Heart J. 2006;27:1841-1846
18. Qu YC, Du YM, Wu SL. et al. Activated nuclear factor-kappaB and increased tumor necrosis factor-alpha in atrial tissue of atrial fibrillation. Scand Cardiovasc J. 2009;23:1-6
19. Adam O, Lavall D, Theobald K. et al. Rac1-induced connective tissue growth factor regulates connexin 43 and N-cadherin expression in atrial fibrillation. J Am Coll Cardiol. 2010;55:469-80
20. Madrid AH, Bueno MG, Rebollo JM. et al. Use of irbesartan to maintain sinus rhythm in patients with long-lasting persistent atrial fibrillation: a prospective and randomized study. Circulation. 2002;106:331-336
21. Ueng KC, Tsai TP, Yu WC. et al. Use of enalapril to facilitate sinus rhythm maintenance after external cardioversion of long-standing persistent atrial fibrillation: results of a prospective and controlled study. Eur Heart J. 2003;24:2090-2098
22. Simm A, Diez C. Density dependent expression of PDGF-A modulates the angiotensin II dependent proliferation of rat cardiac fibroblasts. Basic Res Cardiol. 1999;94:464-471
23. Yoshiharu Higuchi, Kinya Otsu, Kazuhiko Nishida. et al. The Small GTP-binding Protein Rac1 Induces Cardiac Myocyte Hypertrophy through the Activation of Apoptosis Signal-regulating Kinase 1 and Nuclear Factor-B. J Boil Chem. 2003;23:20770-20777
24. Nadruz Jr W, Lagosta VJ, Moreno Jr H, Coelho OR, Franchini KG. Simvastatin prevents load-induced protein tyrosine nitration in overloaded hearts. Hypertension. 2004;43:1060-6
25. Yagi S, Akaike M, Aihara K. et al. Endothelial nitric oxide synthase-independent protective action of statin against angiotensin II-induced atrial remodeling via reduced oxidant injury. Hypertension. 2010;55:918-23
26. Tan AY, Zimetbaum P. Atrial fibrillation and atrial fibrosis. J Cardiovasc Pharmacol. 2011;57:625-9
27. Shiroshita-Takeshita A. et al. Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovas Res. 2007;74:75-84
Author contact
![]() Corresponding author: Yun-qing Chen, M.D, Ph.D. Department of Cardiology, the Second Affiliated Hospital, Chongqing Medical University. No.76 Lingjiang RD., Yuzhong district, Chongqing, 400010 P. R. China. E-mail: chenyunqing.88com or chenyqedu.cn.
Corresponding author: Yun-qing Chen, M.D, Ph.D. Department of Cardiology, the Second Affiliated Hospital, Chongqing Medical University. No.76 Lingjiang RD., Yuzhong district, Chongqing, 400010 P. R. China. E-mail: chenyunqing.88com or chenyqedu.cn.
Received 2013-1-3
Accepted 2013-3-11
Published 2013-4-26