ISSN: 1449-1907International Journal of Medical Sciences
- Current issue
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Volume 18; 2021
- Volume 17; 2020
- Archive
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Rationale of EGFR assessment in...
Pre-treatment EGFR - genotypic...
Conclusions
Abbreviations
Acknowledgements
References

Int J Med Sci 2013; 10(3):320-330. doi:10.7150/ijms.4609 This issue Cite
Review
EGFR-Targeted Therapy for Non-Small Cell Lung Cancer: Focus on EGFR Oncogenic Mutation
Alberto Antonicelli1,5, Stefano Cafarotti2 ![]() , Alice Indini3, Alessio Galli4, Andrea Russo6, Alfredo Cesario7, Filippo Maria Lococo8, Patrizia Russo9, Alberto Franco Mainini5, Luca Giuseppe Bonifati5, Mario Nosotti1, Luigi Santambrogio1, Stefano Margaritora8,10, Pierluigi Maria Granone8, André Emanuel Dutly2
, Alice Indini3, Alessio Galli4, Andrea Russo6, Alfredo Cesario7, Filippo Maria Lococo8, Patrizia Russo9, Alberto Franco Mainini5, Luca Giuseppe Bonifati5, Mario Nosotti1, Luigi Santambrogio1, Stefano Margaritora8,10, Pierluigi Maria Granone8, André Emanuel Dutly2
1. Thoracic Surgery and Lung Transplantation Unit, Foundation IRCCS (Scientific Institute for Research Hospitalization and Health Care) “Ca' Granda” General Hospital, University of Milan, Milan, Italy.
2. Thoracic Surgery EOC Unit - San Giovanni Hospital, Bellinzona, Switzerland.
3. Pediatric Oncology Unit, Foundation IRCCS National Cancer Institute (NCI), Milan, Italy.
4. Department of Preclinical Science LITA Vialba, Luigi Sacco Hospital, University of Milan, Milan, Italy.
5. General Thoracic Surgery Unit, Saronno Regional Hospital, Saronno, Italy.
6. Institute of Anestesiology and Intensive Care Medicine, Catholic University of Sacred Heart, Rome, Italy.
7. Deputy Scientific Director, IRCCS San Raffaele Group, Rome, Italy.
8. Division of Thoracic Surgery, Catholic University of Sacred Heart, Rome, Italy.
9. Laboratory of Systems Approaches and Non Communicable Diseases, IRCCS “San Raffaele Pisana”, Rome, Italy.
10. CFO - Florence Cancer Center, Sesto Fiorentino, Italy.
Abstract
The two essential requirements for pathologic specimens in the era of personalized therapies for non-small cell lung carcinoma (NSCLC) are accurate subtyping as adenocarcinoma (ADC) versus squamous cell carcinoma (SqCC) and suitability for EGFR molecular testing, as well as for testing of other oncogenes such as EML4-ALK and KRAS. Actually, the value of EGFR expressed in patients with NSCLC in predicting a benefit in terms of survival from treatment with an epidermal growth factor receptor targeted therapy is still in debate, while there is a convincing evidence on the predictive role of the EGFR mutational status with regard to the response to tyrosine kinase inhibitors (TKIs).
This is a literature overview on the state-of-the-art of EGFR oncogenic mutation in NSCLC. It is designed to highlight the preclinical rationale driving the molecular footprint assessment, the progressive development of a specific pharmacological treatment and the best method to identify those NSCLC who would most likely benefit from treatment with EGFR-targeted therapy. This is supported by the belief that a rationale for the prioritization of specific regimens based on patient-tailored therapy could be closer than commonly expected.
Keywords: EGFR targeted therapy, NSCLC, advanced, mutation, TKIs, resistance.
Introduction
Alterations in receptor tyrosine kinases (RTKs) including over expression, amplification or mutation have shown to play a key role in the pathogenesis of lung cancer1. In recent years, attention has been paid to the role that “driver mutations” have in tumorigenesis, in order to use them as potential targets for therapy. Such “driver mutations” include those of the epidermal growth factor receptor (EGFR) and of the anaplastic lymphoma kinase (ALK) 2,3.
The EGFR family of TKs, referred to as the HER or ErbB family, consists of four members - EGFR (HER1/ErbB1), HER2 (ErbB2), HER3 (ErbB3) and HER4 (ErbB4). These members regulate many physiological processes and are involved in the modulation of cell proliferation, apoptosis, cell motility and neovascularisation, thus being able to induce important mechanisms related to cancerogenesis4,5. The EGFR tyrosine kinase works through the auto-activation of the receptor via its homo/heterodimerization and autophosphorylation on tyrosine-rich cytosolic domains after the binding of the ligand. This leads to the beginning of two main downstream intermediate pathways: the PIK3CA/AKT1/MTOR pathway and the RAS/RAF1/MAP2K1/MAPK1 kinases6. There is evidence that the activated EGFR can also mediate signals through the STAT transcription factors7,8. Improper activation and over-expression of EGFR-TK results in increased cell proliferation, survival, invasion and metastasis. This has been implicated in the pathogenesis and progression of many malignancies as well as in the poor prognosis of patients 7,9-10. In malignant cells, including NSCLC cells, the activity of the receptor may become dysregulated and no longer under the control of inherent inhibitory mechanisms11.
Spontaneous EGFR mutations often are oncogenic; that is, they activate the EGFR-signalling pathway in the absence of ligand and promote cell proliferation, survival and anti-apoptotic signals. These signalling networks make EGFR-mutated cells dependent on a functional EGFR for their survival, rendering them addicted to the receptor. Inhibition of EGFR leads to up-regulation of pro-apoptotic molecules and finally results in cell death through the activation of the intrinsic mitochondrial apoptotic pathway12,13. There are several described mutations in the EGFR gene. The two most common are: 1) short in-frame deletions around the LREA motif of exon 19 (~45-50% of mutations); and 2) a point mutation (CTG to CGG) in exon 21 that results in substitution of leucine by arginine at codon 858, L858R (~45-50% of mutations)14,15. These mutations are more frequently found in NSCLC with an adenocarcinoma histology, tumors in women, East Asians and never smokers14-16.
EGFR mutations in lung cancers constitute one of the major subsets among those molecular aberrations occurring in lung cancers. The incidence of EGFR mutations in tumors with non-small-cell histology ranges from ~15% in Caucasians to ~50% in East Asians17; 95% of such mutations have been found in adenocarcinomas18. Patients bearing EGFR mutations have shown favourable clinical outcomes even with conventional chemotherapy suggesting that EGFR may serve as a predictive factor as well as a prognostic factor19.
Over 50% of patients diagnosed with NSCLC present with stage IIIB or IV disease is not amenable to curative treatment20 and the only pathologic material guiding systemic therapy may be small biopsy and cytology specimens.
Until the recent use of TKIs, the standard first-line treatment for most patients with unresectable NSCLC and good performance status has involved the use of a combination of chemotherapy regimens (usually cisplatin-based), which from the 1970s and 1980s were shown to reproducibly achieve objective response in 20% to 30% of advanced NSCLC patients. The most common combination regimens in use at present are gemcitabine with either cisplatin or carboplatin, followed by paclitaxel-carboplatin, vinorelbine-platinum and docetaxel-platinum combinations21,22. The addition of the recombinant humanized monoclonal antibody bevacizumab that binds to vascular endothelial growth factor (VEGF) to carboplatin and paclitaxel for the treatment of non-squamous advanced NSCLC has demonstrated to increase RR, PFS and OS when compared to chemotherapy alone23. Disease progression affects almost all patients after initial treatment and requires additional therapy. The agents approved for second-line therapy in advanced NSCLC are docetaxel24, pemetrexed25 and erlotinib26. When tested in randomized trials24-26, these agents have demonstrated a PFS below 2-3 months with a median overall survival no longer than 9 months in very few unselected patients.
Despite recent advances with approval of more active chemotherapeutic and anti-angiogenesis agents for stage IV NSCLC, standard therapy can provide only modest clinical benefits with significant toxicities when used in unselected patients. In 2004, the identification of somatic mutations in the EGFR gene provided the first glimpse of a possible target for a treatment27,28 which could maximize clinical outcome in those patients who could benefit from a personalized therapy29. This implies the identification of certain characteristic molecular lesions meant to be causally responsible for maintenance of the malignant phenotype and also distinctive of the cancer cells. Therapies targeted to these molecular lesions offer the prospect for tumor control and selectivity with less toxicity than traditional chemotherapy.
This review is designed to shed light on the rationale supporting the preclinical molecular footprint assessment, on the progressive development of specific pharmacological treatments and on the best method to identify those NSCLCs which would most likely benefit from treatment with EGFR-targeted therapy.
Rationale of EGFR assessment in Non Small Cell Lung Cancer
Somatic mutations in the EGFR gene which account for oncogenesis in NSCLCs are activating mutations that work through a “gain of function” mechanism (i.e. the ligand-independency of the tyrosine kinase's signalling activity). These types of mutations can be found in 10 to 20% of patients with a NSCLC at an advanced stage and in more than 50% of adenocarcinomas and tumours from East Asians, never smokers and women30. The described mutations generally interest the exons of the EGFR gene between the 19th and the 21th, which correspond to the portion of the receptor with kinase activity, nearby the binding site of ATP (that is fundamental to the activation of the receptor by autophosphorylation). As a result, the receptor is blocked into a state of constitutive activation, signalling to the cell to proliferate and to resist apoptosis. The two most frequent genic mutations responsible for this anomaly in cell cycle are the substitution of arginine for leucine at codon 858 (L858R), exon 21, and in-frame deletions at exon 19 (del E746_A750 is the most common). L858R accounts for 45-50% of mutations while deletions at exon 19 account for another 45-50%. Other rarer mutations (5%) associated with EGFR constitutive activation are insertions in exon 20 and substitutions at the glycine residue at codon 719 in exon 18 (as the G719S mutation). Molecular studies on the receptors harbouring a known L858R mutation demonstrated a decreased affinity of the tyrosine kinase for ATP and an increase in the affinity for the tyrosine kinase inhibitors (TKI). These are drugs studied to specifically target the pathways of oncogenesis mediated by the EGFR hyper-activation and causing little harm to non-tumoral cells30. There is evidence supporting a different sensitivity of NSCLCs to the TKI gefitinib depending on the presence of an exon 21 mutation or an exon 19 deletion, having deletions at exon 19 been associated with a better response31.
Furthermore, in about 30-75% of samples from NSCLCs, an EGFR over-expression30 due to epigenetic causes (transcriptional hyper-activation), gene amplification or oncogenic viruses can be detected7. EGFR over-expression and the consequent augmentation of cell survival and proliferation has also been observed in premalignant lesions, showing the importance of this membrane-associated receptor in tumor growth7. NSCLCs that over-express both EGFR and HER2 (another member of the HER family) demonstrated an aggressive tumor cell growth. In effect, EGFR/HER2 heterodimers display a greater activity than EGFR homodimers7. Some studies demonstrated that the EGFR gene amplification, and thus over-expression directly correlates with a better response to tyrosine kinase inhibitors, and NSCLCs that over-express both EGFR and HER2 are more sensitive to this class of drugs than a tumor with an increased expression of EGFR alone. In this perspective, pre-treatment assessment of EGFR and HER2 gene copy number may have a prognostic and a predictive value of response to TKIs in NSCLCs7,32, but there are no univocal opinions with regard to this topic. However, data from the recent phase 3 FLEX study on patients with advanced NSCLC underpin pre-treatment assessment of EGFR expression by IHC, for EGFR over-expression on the surface of tumoral cells proved to be predictive of better overall survival with a combined treatment of first-line chemotherapy (comprising cisplatin and vinorelbine) plus cetuximab (an anti-EGFR monoclonal antibody) relative to chemotherapy alone. Among patients with a high EGFR expression, those who were in the combined treatment group had a median OS of 12 months versus the 9.6 months of patients allocated to the chemotherapy alone arm33. Which strategy is better in the management of advanced NSCLCs, if the inhibition of mutated EGFR with TKIs or a combined approach with cetuximab plus first-line chemotherapy, is yet to be determined. The combination of cetuximab and a tyrosine kinase inhibitor in the treatment of those tumors which are resistant to TKIs alone is intriguing34,35, but to date clinical data are still lacking.
EGFR expression seen in advanced lung cancers made this an attractive target for molecular intervention in this disease. Several studies (Table 1) have by now demonstrated dramatic improvement in response rates, quality of life, symptoms, and median progression-free-survival (by 2-5 months) with first-line EGFR-TKI therapy compared with standard platinum-doublet chemotherapy in patients with EGFR mutation-positive NSCLC36,37. Gefitinib and erlotinib were the first two agents to target the tyrosine kinase domain of the EGFR. Both these agents showed encouraging activity in patients with NSCLC who had been previously treated with chemotherapy in the phase I series and then in phase II trials36,37. This has led to their approval of the treatment in advanced NSCLC. The first two large phase III trials comparing EGFR TKIs in patients previously treated with a different first line therapy were BR.2138 and INTEREST39 trials. BR21 was an international phase III randomized trial comparing erlotinib with placebo in patients who had failed at least one line of cytotoxic chemotherapy. With the survival benefit shown in this trial, erlotinib has been approved for use as second or third-line therapy in NSCLC after failure with a cytotoxic chemotherapy regimen by American and European regulatory agencies38. A randomised, open-label phase III trial (INTEREST)39 of gefitinib versus docetaxel in previously treated patients established non-inferior survival in patients treated with TKIs compared with chemotherapy, thus suggesting that gefitinib is a valid treatment for pre-treated patients with non-small-cell lung cancer39.
In contrast to the significant clinical and radiological responses seen in patients harbouring EGFR activating mutations, gefitinib and erlotinib have shown only limited activity in non-EGFR genotyped, or unselected, NSCLCs when given as first, second or subsequent lines of therapy.37,40. This has been reported by several prospective trials of gefitinib and erlotinib in EGFR-mutated NSCLC, which showed RRs exceeding 70% in tumors with exon 19 deletions or the L858R mutation, with PFS intervals of 6-14 months and OS times beyond 20-24 months40-43.
During the last three years, the predictive value of EGFR mutations for use of gefitinib has been strengthened by the results of three randomized phase III trials that specifically compared TKIs used as first-line therapy with traditional platinum-based chemotherapy in patients with advanced NSCLC. In 2009 the results of IRESSA Pan-Asia Study36,44 were presented. This trial included 1217 patients of Asian ethnicity who were never smokers or former light smokers yet had histologic diagnosis of adenocarcinoma. The trial demonstrated an improvement in PFS and RR (with no statistical difference in OS) with the use of gefitinib in EGFR-mutated tumors and, in contrast, better RR and PFS with standard chemotherapy in patients without mutations. The first phase III trial of gefitinib versus chemotherapy as initial treatment of recurrent or advanced NSCLC, based on selection of patients with known activating EGFR mutations was the WJTOG3405 trial, reported in 201045. This trial documented important achievements in terms of RR and PFS with the use of TKIs. During the same year, such results were confirmed by another similar Japanese phase III trial, NEJ00237, with RR and PFS definitely favouring the use of gefitinib in the first-line setting of metastatic EGFR-mutated NSCLC.
Summary of first-line efficacy data of TKIs in patients with EGFR-mutated advanced NSCLC (results from randomized phase III trials of gefitinib/erlotinib versus standard chemotherapy).
| Trial | patient selection | treatment/number of patients | ORR (%) | PFS (months) | OS (months) |
|---|---|---|---|---|---|
| IPASS | Asia | Carboplatin-Paclitaxel | 47 | 6.3 | 21.9 |
| (Mok et al. 2009) | never- or light ex-smoker | (n=608 total; n=129 EGFR M+) Gefitinib | 71 | 9.5 | 21.6 |
| adenocarcinoma | (n=609 total; n=132 EGFR M+) | p < 0.001 | HR 0.48; p<0.001 | HR 1.00; p 0.99 | |
| WJTOG 3405 | Asia | Cisplatin-Docetaxel | 32 | 6.3 | not reached |
| (Mitsudomi et al. 2010) | EGFR mutation | (n=86 EGFR M+) Gefitinib | 62 | 9.2 | 30.9 |
| (n=86 EGFR M+) | p< 0.001 | HR 0.49; p<0.0001 | HR 1.64; p 0.211 | ||
| NEJ 002 | Asia | Carboplatin-Paclitaxel | 31 | 5.4 | 23.6 |
| (Maemondo et al. 2010) | EGFR mutation | (n=100 EGFR M+) Gefitinib | 74 | 10.8 | 30.5 |
| (n=98 EGFR M+) | p< 0.001 | HR 0.30; p<0.001 | HR NR; p 0.31 | ||
| OPTIMAL | Asia | Carboplatin-Gemcitabine | 36 | 4.6 | NA |
| (Zhou et al. 2011) | EGFR mutation | (n=72 EGFR M+) Erlotinib | 83 | 13.1 | NA |
| (n=83 EGFR M+) | p< 0.0001 | HR 0.16; p<0.0001 | NA | ||
| EURTAC | Europe | Platinum-Gemcitabine/Docetaxel | 15 | 5.2 | 19.5 |
| (Rossell et al. 2012) | EGFR mutation | (n=87 EGFR M+) Erlotinib | 58 | 9.7 | 19.3 |
| (n=86 EGFR M+) | OR 7.5; p<0.0001 | HR 0.37; p<0.0001 | HR 1.047; p 0.87 |
EGFR: epidermal growth factor receptor; ORR: objective response rate; PFS: progression-free survival; OS: overall survival; HR: hazard ratio; OR: odds ratio; NA: not available, NR: not reported.
Numerous small studies (mainly conducted in East-Asia) on EGFR-TKI monotherapy with gefitinib rapidly confirmed high objective response rate with this agent used in first-line setting in patients with cancers harbouring a mutation42,43,46-49. Based on the results of the IPASS study, gefitinib was approved for use in Europe for the initial treatment of patients with NSCLC exhibiting EGFR mutations. Confirmatory randomized phase III trials of erlotinib versus standard chemotherapy have recently been concluded in Asia (OPTIMAL trial, NCT0087441950) and Europe (EURTAC trial, NCT0044622551). The positive results of these studies suggested that responsiveness in mutation-positive patients was not a function of ethnicity. Furthermore, Caucasian patients demonstrated a spectrum of EGFR mutational subtypes similar to those seen in East Asian patients. Gefitinib and erlotinib have shown a similar spectrum of activity, with little differences in pharmacokinetics determining a major bioavailability for erlotinib52. This is the only TKI which has been approved by FDA for the management of treatment-naive patients with advanced NSCLC showing EGFR activating mutations53.
EGFR-TKIs as a class are generally well tolerated. The two most common toxicities include dermatologic and GI effects; both of which are mild to moderate, easily managed and reversible36,37,54.
In order to determine whether an EGFR TKI or chemotherapy is the appropriate first-line therapy, the latest guidelines55 recommend mutation testing for all patients with advanced NSCLC tumor.
All EGFR-mutated patients treated with gefitinib or erlotinib invariably develop acquired resistance to this kind of therapy56,57 (Figure 1). The most common and first identified mutation is the threonine-790 to methionine (T790M) point mutation in exon 20 which represents approximately 50% of all acquired resistance in NSCLC58. The development of such genetic alteration restores the EGFR TK affinity to ATP, rendering first-generation TKIs inactive59,60. Other secondary resistance mutations within the same gene have been reported infrequently (L747S, D761Y, T854A)12,61-62. All these mutations, together with T790M, have also been identified in pre-treatment tumors and, similarly, are responsible for both a lesser sensitivity and duration of response to the first generation TKIs61,63-65. Other mechanisms of acquired resistance include MET gene amplification (also accounting for up to 20% of pre-treatment tumoral resistances)66, increased signalling through parallel pathways such as the ones of VEGF67 and IGF1R68, mutations and activation of PIK3CA69,70 and transformation into a small-cell lung cancer phenotype71.
Concerns have been raised about resistance to TKIs due to mutations in the KRAS gene (which encodes for a protein downstream to the EGFR in the pathway initiated by the activation of the tyrosine kinase) or due to mutations that occur in other proteins of the same cascade (e.g. BRAF72). Even if some studies have underlined that KRAS mutations are negative predictors of sensitivity to TKIs in NSCLCs73, more recent research demonstrates that they are not necessarily associated with a poor response to this receptor-targeted therapy; this is contrary to the absence of EGFR mutations, which accounts for the lack of effectiveness of treatment with tyrosine kinase inhibitors74,75.
Management of EGFR tumor resistance has become the next challenge in order to lengthen these patients' overall survival; identification of the molecular resistance mechanisms will allow for the treatment of TKI-resistant tumors. A new class of drugs, the so-called second-generation TKIs, may be able to overcome the T790M mutation resistant cell. Compared to first-generation TKIs, these molecules show higher affinity for the ATP-binding domain, form an irreversible covalent bond to the ATP-binding site and are able to stimulate other receptors (e.g. HER2). Neratinib (HKI-272), one of the three agents investigated, hasn't shown good RR when tested on patients with known T790M mutation76, therefore further development of this drug in lung cancer has been halted. Afatinib (BIBW2992) is being investigated as part of the LUX-Lung program which aims to evaluate the use of TKI in second- or third-line treatment in patients who have acquired resistance to gefitinib or erlotinib (LUX-Lung 1, 4 and 5) as well as the use of TKIs as a first-line treatment in patients with EGFR-activating mutations (LUX-Lung 2, 3 and 6). Therapy with afatinib has demonstrated to improve the disease control rate and to prolong PFS in both LUX-Lung 1 and 277,78. The G719S mutation renders the tumor less sensitive to gefitinib, while erlotinib and the second-generation TKI afatinib have proven to be effective in tumors characterized by this substitution30. Dacomitinib (PF-00299804) is another irreversible TKI able to target the activity of all HER TKs and has shown activity in NSCLC cell lines harbouring the T790M mutation79. This molecule has been evaluated in two phase II trials: the first one was after failure of one or two chemotherapy regimens and failure on erlotinib80; the second one compared it with erlotinib in the second- and third-line in patients with advanced NSCLC81. The results of these studies seem promising and further studies will evaluate dacomitinib in upfront therapy over first-generation TKIs and in resistant EGFR-mutated tumors82.
Pre-treatment EGFR - genotypic assessment
As the EGFR mutational profile of NSCLCs is a strong predictor of response to therapy with the highly effective TKIs36,37, the most recent algorithms for the management of advanced NSCLCs underline the importance of EGFR molecular testing prior to the initiation of therapy30,83, adopting either a traditional Sanger's sequencing-based analysis30,84 or PCR assays30. Recent advances in genome analysis technologies lead to the use of high-throughput next generation sequencing platforms, which may become more available in routine clinical practice in a near future85,86. In particular, EGFR mutations should be sought in those NSCLCs in which they occur most frequently: adenocarcinomas, NSCLCs in people who have never smoked, Asians and women30. A first-line treatment with TKIs should be initiated in those patients who have a positive result for EGFR mutations, otherwise a standard chemotherapy should be considered30,83. EGFR mutation-positive NSCLCs which also test positive for mutations associated with primary or, most frequently, secondary resistance to first-generation TKIs (first of all the T790M substitution) may respond better to newer agents like afatinib, and dacomitinib, which have demonstrated a greater effectiveness in these clinical situations, but more clinical data are required30,56,87-89.
Ways to leave your EGFR inhibitor: Biochemical pathways leading to resistance to small molecule EGFR drugs such as gefitinib and erlotinib. (A) Structures of two approved EGFR TKIs, gefitinib and erlotinib, used in the treatment of NSCLC. (B) Ribbon diagram of wild-type human EGFR (PDB code 2ITY), illustrating binding of gefitinib to the active site of the kinase. The magenta ball-stick (located just above the gefitinib molecule in the active site) indicates the gatekeeper residue (threonine790) that is commonly mutated to methionine (T790M), resulting in reduced inhibitor binding and drug resistance. (C) Simplified pathway diagram of EGFR signaling through RAS/MEK/ERK and PI3K/PDK1/AKT indicating the points of mutation/amplification in EGFR TKI resistance as reported by Sequist and colleagues. The resistance mechanisms include the EGFR T790M gatekeeper mutation, amplification of EGFR T790M, MET amplification, and PI3KCA mutation (note that additional epithelial to mesenchymal transition changes and transformation from the NSCLC to the SCLC phenotype also lead to resistance but are not covered by this illustration). The illustration also shows the FAS/NF-kB signaling arm downstream of the FAS death receptor that was shown to be important in TKI resistance by Bivona and colleagues. Reprinted from Cancer Cell, 19, Paul Workman and Paul A. Clarke, “Resisting Targeted Therapy: Fifty Ways to Leave Your EGFR”, 437-440, 2011, with permission from Elsevier.
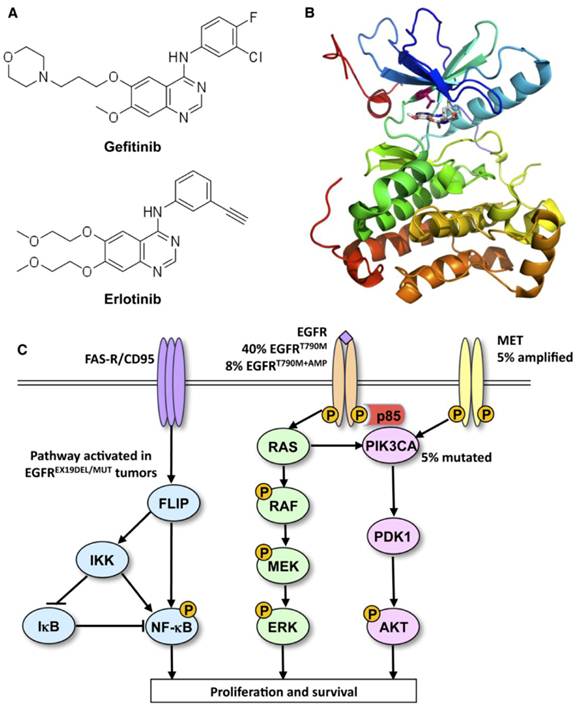
Furthermore, some studies have supported the validity of pre-treatment measurement of EGFR gene copy number by FISH in predicting response to TKIs7,90-92. NSCLCs which over-express both EGFR and HER2 (over-expression directly correlates with FISH measurement of gene copy number) seem to be more sensitive to TKIs, providing another criterion that can help the clinician in therapeutic decision making7,32. In any case, these findings on the role of tyrosine kinase receptors' over-expression as a positive predictor of response to molecular-targeted drugs are controversial because of the conflicting evidence of different trials55,90-92. It is worth noting that the conflicting evidence may also be due to methodological differences among laboratories performing the FISH assay or to different interpretations of FISH results90.
Accordingly, the only strong evidence on the effectiveness of predictors of success in the treatment of advanced NSCLCs with TKIs is in favor of EGFR mutation assessment55,90-92.
In future, when other classes of now developmental targeted drugs are available in clinical practice, the assessment of mutations or overexpression of genes other than EGFR in EGFR negative NSCLCs (KRAS, HER2, ALK/EML4-ALK, PIK3CA, BRAF, FGFR1) may become routine30,83.
Although genotypic assessment may be useful in determining which patients are likely to respond to EGFR TKI therapy, problems are associated with obtaining suitable DNA for pre-treatment analysis.
The quality of the samples and the extracted DNA and the quantity of DNA available using current methods may limit the routine use of genotypic assessment. As described above, cancer cells from various sources have been used for genotyping. These include archived surgically resected tissue from patients who subsequently develop recurrent or progressive disease, formaline-fixed paraffine-embedded biopsy tissue, frozen sections, cytology specimens from lavage, pleural effusion, FNA (CTCs), tumor DNA in serum and tumor DNA in plasma or blood. The mutation status of EGFR can be also determined in tumor specimens obtained during curative surgery or at biopsy. Surgery offers the best chance of high-quality and high-volume tumor tissue samples, however only 20-25% of lung cancers are suitable for curative surgery and EGFR TKI therapy is only licensed for use in patients with advanced disease.
Generally, there is only a small amount of tumor in routine diagnostic biopsy samples from patients with advanced disease; that is why specimens should be optimized and handled properly so that the most accurate pathologic characterization is achieved by individualizing among NSCLC, especially in adenocarcinoma, those histotypes that are more frequently driven by EGFR mutation. Especially for poorly differentiated neoplasms, a significant amount of specimen is often needed for diagnostic purposes and “consumed” for special immunohistochemical stains, as P63, P40 (markers of squamous differentiation), TTF-1 (positive in most of primitive lung adenocarcinoma), cromogranine and synaptophysine (marker of neuroendocrine differentiation), therefore reducing the material available for molecular testing. Lim and colleagues were able to obtain sufficient genomic DNA for genotypic assessment from more than 80% of their 24 low-volume samples (needle or forceps biopsy or fine-needle aspiration)93. Of the 139 patients studied by Shih and colleagues94 only two had insufficient DNA for analysis, whereas Savic and colleagues95 successfully sequenced the DNA from 93% of their 84 cytological NSCLC specimens. Nakajima and colleagues96 determined EGFR mutation status in all 43 patients in their study, and Yoshida and colleagues97 determined EGFR mutation status in all 35 fine-needle aspiration/biopsy samples in their study.
What is of crucial importance is that any tested sample is checked for adequacy in terms of the number and percentage of tumor cells. Two recently published studies reporting successful EGFR mutation detection from FNA cytology samples found EGFR mutation rates significantly lower than those reported from the same laboratories using tissue biopsies98,99, while a recent review reported a sensitivity between 92% and 100% for EGFR mutation detection in tissue samples, varying with different techniques, and comparable results with cytology samples100.
Follow-up biopsy to determine the T790M mutation status may be unnecessary if further research supports the initial finding that DNA sufficient for genotypic assessment can be isolated from blood samples101-103. A study reported a 92% sensitivity for detection of EGFR mutations in patients with metastatic NSCLC with this method (11 of 12 patients)83.
There seems to be homogeneity of EGFR mutational state within the same tumoral lesion, even if histologically heterogeneous, and this provides a rationale for a sample-driven therapy104. However, it remains to be further elucidated if there are practical consequences in relation to the finding that NSCLCs' metastases in extrapulmonary organs or multiple intrapulmonary nodules can either differ from the primary lesion or from each other in regards to the EGFR mutational state105-107 and expression107 as detected by the laboratory. It is in this context if multiple biopsies are necessary to correctly evaluate the EGFR profile. Moreover, the response of some EGFR mutation positive patients to TKIs treatment has proven to be "mixed"105: this means that clinical responses which are not completely consistent with those expected from the EGFR mutation analysis, could be explained by the aforementioned differences in the EGFR mutational state and expression among different tumoral sites.
Furthermore, it seems that chemotherapy is able to modify the EGFR expression in NSCLCs107 by increasing or decresing it. This should be taken into consideration when anti-EGFR monoclonal antibody cetuximab is used to treat patients whose tumoral EGFR expression was assessed before chemotherapy: in this case, EGFR expression should be reassessed after having performed chemotherapy.
Conclusions
It is reasonable to suggest that personalized therapy for NSCLC patients should include a genetic assessment of the EGFR mutational status for individual patients. In this scenario, our overview aims to focus on the necessity of standardizing and improving pre-diagnostic and diagnostic tools and of optimizing the accuracy and sensitivity of EGFR mutational testing so that it might be introduced into routine clinical practice. The appropriate role of an EGFR mutation routine analysis in the treatment of patients with NSCLC continues to evolve; this on the basis of new prospective clinical studies providing new standards of care such as adequate documentation of the EGFR mutational status in the preclinical setting, during the treatment and in related follow-up.
There is reasonable basis to believe in the use of instrumental molecular reassessment of neoplastic biological characteristics for patients with advanced NSCLC throughout the clinical course in order to finely tune the treatment, particularly in the case of disease progression. In this way, TK inhibitors could be used for the best outcome in the patient, and the drug resistance that supervenes during treatment can thus be promptly identified. In addition, close cooperation between clinicians, surgeons, molecular biologists and pathologists is crucial for a continuous improvement in the field of NSCLC target therapy.
Abbreviations
NSCLC: non small cell lung cancer; ADC: adenocarcinoma; SqCC: squamous cell carcinoma; RTKs: receptor tyrosine kinases; TKIs: tyrosine kinase inhibitors; ALK: anaplastic lymphoma kinase; HER: human epidermal growth factor receptor; ErbB: erythroblastic leukemia viral oncogene homolog; PIK3CA: phosphatidylinositol 3′-kinase; AKT: protein kinase B (PKB); mTOR: mammalian target of rapamycin; RAS: rat sarcoma; RAF: rapidly accelerated fibrosarcoma; MAP: mitogen-activated protein; STAT: signal transducers and activators of transcription; VEGF: vascular endothelial growth factor; RR: response rate; ATP: adenosine-5'-triphosphate; IPASS: Iressa pan-Asia study; EURTAC: European Tarceva Versus Chemotherapy; FDA: food and drug administration; GI: gastrointestinal; MET: MNNG HOS Transforming gene; IGF1R: insulin-like growth factor 1 receptor; KRAS: v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; BRAF: v-raf murine sarcoma viral oncogene homolog B1; PCR: polymerase chain reaction; IHC: immunohistochemistry; FISH: fluorescence in situ hybridization; FGFR1: fibroblast growth factor receptor 1; DNA: deoxyribonucleic acid; FNA: fine-needle aspiration; CTCs: circulating tumor cells; SCLC: small cell lung cancer.
Acknowledgements
The authors gratefully thank Dr. Andrea Vingiani, resident in Anatomic and Clinical Pathology at the University of Milan, for his sincere cooperation.
We would like to dedicate the efforts of all our work to Professor Laura Vizzotto.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arteaga C. Targeting HER1/EGFR: a molecular approach to cancer therapy. Semin Oncol. 2003;30:3-14
2. Paez JG, Janne PA, Lee JC. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2002;304:1497-1500
3. Lynch TJ, Bell DW, Sordella R. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib therapy. N Engl J Med. 2004;350:2129-2139
4. Cheng L, Zhang S, Alexander R. et al. The landscape of EGFR pathways and personalized management of non-small-cell lung cancer. Future Oncol. 2011;7:519-541
5. Normanno N, De Luca A, Bianco C. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2-16
6. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341-354
7. HirschFR Varella-Garcia M, Capuzzo. Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene. 2009;28:S32-S37
8. Quesnelle KM, Boehm AL, Grandis JR. STAT-mediated EGFR signaling in cancer. J Cell Biochem. 2007;102(2):311-9
9. Wang X, Zhang S, MacLennan GT. et al. Epidermal growth factor receptor protein expression and gene amplification in the chemorefractory metastatic embryonal carcinoma. Mod Pathol. 2008;22:7-12
10. Wang X, Zhang S, MacLennan GT. et al. Epidermal growth factor receptor protein expression and gene amplification in small cell carcinoma of the urinary bladder. Clin Cancer Res. 2007;13:953-957
11. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367-1380
12. Costa DB, Halmos B, Kumar A. et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669-1679
13. Sharma SV, Haber DA, Settleman J. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169-181
14. Sequist LV, Bell DW, Lynch TJ. et al. Molecular predictor of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J ClinOncol. 2007;25:587-595
15. Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21(Suppl 2):S16-S22
16. Shigematsu H, Lin L, Takahashi T. et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancer. J Natl Cancer Inst. 2005;97:339-346
17. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. The Lancet Oncology. 2011;12:175-180
18. Yamamoto H, Toyooka S, Mitsudomi T. Impact of EGFR mutation analysis in non-small-cell lung cancer. Lung Cancer. 2009;63:315-321
19. Eberhard DA, Johnson BE, Amler LC. et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J ClinOncol. 2005;23:5900-5909
20. Schrump DS, Altorki NK, Henschke CL. et al. Non-small cell lung cancer. In: (ed.) DeVita VT. Cancer: Principles and Practices of Oncology. Philadelphia: Lippincott Williams & Wilkins. 2005:753-810
21. Azzoli CG, Baker Jr S, Temin S. et al. American Society of Clinical Oncology Clinical Practice Guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol. 2009;27:6251-6266
22. Fossella F, Pereira JR, von Pawel J. et al. Randomized, multinational phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J ClinOncol. 2003;21:3016-3022
23. Sandler A, Gray R, Perry MC. et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542-2550
24. Shepherd FA, Dancey J, Ramlau R. et al. Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol. 2000;18:2095-2103
25. Hanna N, Shepherd FA, Fossella FV. et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J ClinOncol. 2004;22:1589-1597
26. Shepherd FA, Rodrigues PJ, Ciuleanu T. et al. Erlotinib in previously treated non-small-cell lung cancer. NEngl J Med. 2005;353:123-132
27. Lynch TJ, Bell DW, Sordella R. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-2139
28. Paez JG, Janne PA, Lee JC. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497-1500
29. Fujita S, Katakami N, Masago K. et al. Customized chemotherapy based on epidermal growth factor receptor mutation status for elderly patients with advanced non-small-cell lung cancer: a phase II trial. BMC Cancer. 2012;12:185
30. Soria JC, Mok TS, Cappuzzo F, Pasi AJ. EGFR-mutated oncogene-addicted non-small cell lung cancer: Current trends and future prospects. Cancer treatment reviews. 2012;38:416-430
31. Zhu J, Zhong W, Zhang G. Better survival with EGFR exon 19 than exon 21 mutations in gefitinib-treated non-small cell lung cancer patients is due to differential inhibition of downstream signals. Cancer Lett. 2008;265(2):307-17
32. Cappuzzo F, Varella-Garcia M, Shigematsu H. et al. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. Journal of Clinical Oncology. 2005;23:22
33. Pirker R, Pereira JR, von Pawel J. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012;13:33-42
34. Wang M, Zhao J, Zhang LM. Combined Erlotinib and Cetuximab overcome the acquired resistance to epidermal growth factor receptors tyrosine kinase inhibitor in non-small-cell lung cancer. J Cancer Res Clin Oncol. 2012;138(12):2069-77
35. Kim HP, Han SW, Kim SH. Combined lapatinib and cetuximab enhance cytotoxicity against gefitinib-resistant lung cancer cells. Mol Cancer Ther. 2008;7(3):607-15
36. Mok TS, Wu YL, Thongprasert S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947-957
37. Maemondo M, Inoue A, Kobayashi K. et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380-2388
38. Zhu CQ, da Cunha Santos G, Ding K. et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J ClinOncol. 2008;26:4268-4275
39. Kim ES, Hirsh V, Mok T. et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008;372:1809-1818
40. Costa DB, Kobayashi S, Tenen DG. et al. Pooled analysis of the prospective trials of gefitinib monotherapy for EGFR-mutant non-small-cell lung cancer. Lung Cancer. 2007;58:95-103
41. Rosell R, Moran T, Queralt C. et al. Screening for epidermal growth factor mutations in lung cancer. N Engl J Med. 2009;361:958-967
42. Sequist LV, Martins RG, Spigel D. et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442-2449
43. Tamura K, Okamoto I, Kashii T. et al. Multicentre prospective phase II trial of gefitinib for advanced non-small-cell lung cancer with epidermal growth factor receptor mutations: results of the West Japan Thoracic Oncology Group trial (WJTOG0403). Br J Cancer. 2008;98:907-914
44. Mok T, Wu YC, Thongprasert S. et al. Phase III, randomised, open-label, first-line study gefitinib (G) vs carboplatin/paclitaxel (C/P) in clinically selected patients (PTS) with advanced non-small-cell lung cancer (NSCLC) (IPASS). Ann Oncol. 2008;19:AbstractLBA2
45. Mitsudomi T, Morita S, Yatabe Y. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121-128
46. Inoue A, Suzuki T, Fukuhara T. et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small cell lung cancer with epidermal growth factor mutations. J ClinOncol. 2006;24:3340-3346
47. Asahina H, Yamazaki K, Kinoshita I. et al. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor. Br J Cancer. 2006;95:998-1004
48. Yoshida K, Yatabe Y, Park JY. et al. Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer. J Thorac Oncol. 2007;2:22-28
49. Sugio K, Uramoto H, Onitsuka T. et al. Prospective phase II study of gefitinib in non-small cell lung cancer with epidermal growth factor receptor gene mutations. Lung Cancer. 2009;64:314-318
50. Zhou C, Wu Y-L, Chen G. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735-742
51. Rosell R, Carcereny E, Gervais R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239-246
52. Scheffler M, Di Gion P, Doroshyenko O. et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on 4-anilinoquinazolines. ClinPharmacokinet. 2011;50(6):371-403
53. Ettinger DS, Akerley W, Bepler G. et al. Non-small-cell lung cancer. J NatlComprCancNetw. 2010;8:740-801
54. Thongprasert S, Duffield E, Saijo N. et al. Health-related quality-of-life in a randomized fase III first-line study of Gefitinib versus Carboplatin/Paclitaxel in clinically selected patients from Asia with advanced NSCLC (IPASS). J ThoracOncol. 2011;6:1872-1880
55. Keedy VL, Temin S, Somerfield MR. et al. American Society of Clinical Oncology Provisional Clinical Opinion: Epidermal Growth Factor Receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR Tyrosine kinase inhibitor therapy. J ClinOncol. 2011;29:2121-2127
56. Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10:281-289
57. Jackman D, Pao W, Riely GJ. et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357-360
58. Suda K, Onozato R, Yatabe Y. et al. EGFR T790M mutation: a double role in lung cancer survival?. J Thorac Oncol. 2009;4:1-4
59. Pao W, Miller VA, Politi KA. et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73
60. Yun CH, Mengwasser KE, Toms AV. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. ProcNatlAcadSci USA. 2008;105:2070-2075
61. Bean J, Riely GJ, Balak M. et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519-7525
62. Balak MN, Kwak EL, Shaw AT. et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494-6501
63. Maheswaran S, Sequist LV, Nagrath S. et al. Detection of mutations in EGFR in circulating lung cancer cells. N Engl J Med. 2008;359:366-377
64. Ayoola A, Barochia A, Belani K, Belani C. Primary and acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer: an update. Cancer Invest. 2012;30(5):433-446
65. Su KY, Chen HY, Li KC. et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small cell lung cancer. J Clin Oncol. 2012;30(4):433-440
66. Chen HJ, Mok TS, Chen ZH. et al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non small cell lung cancer. PatholOncol Res. 2009;15:651-658
67. Viloria-Petit A, Crombet T, Jothy S. et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090-5101
68. Guix M, Faber AC, Wang SE. et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609-2619
69. Okudela K, Suzuki M, Kageyama S. et al. PIK3CA mutation and amplification in human lung cancer. PatholInt. 2007;57:664-671
70. Yamamoto H, Shigematsu H, Nomura M. et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68:6913-6921
71. Sequist LV, Waltman BA, Dias-Santagata D. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. SciTransl Med. 2011;3:75ra26
72. Ohashi K, Sequist LV, Arcila ME. et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. ProcNatlAcadSci U S A. 2012;109(31):E2127-33
73. Linardou H, Dahabreh IJ, Kanaloupiti D. et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9(10):962-72
74. Pesek M, Benesova L, Belsanova B. et al. Dominance of EGFR and insignificant KRAS mutations in prediction of tyrosine-kinase therapy for NSCLC patients stratified by tumor subtype and smoking status. Anticancer Res. 2009;29(7):2767-73
75. Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor- targeted therapy?. J Clin Oncol. 2010;28(31):4769-77
76. Sequist LV, Besse B, Lynch TJ. et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of phase II trial in patients with advanced non small-cell lung cancer. J Clin Oncol. 2010;28:3076-3083
77. Miller VA, Hirsch V, Cadranel J. et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528-538
78. Yang C, Shih J, Su W. et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13:539-548
79. Engelman JA, Zejnullahu K, Gale CM. et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancers models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924-11932
80. Boyer MJ, Blackhall FH, Park K. et al. Efficacy and safety of PF00299805 versus erlotinib: a global, randomised phase 2 trial in patients with advanced non small cell lung cancer after failure of chemotherapy. In: Proceedings of 46th Annual Meeting of the American Society of Clinical Oncology. J Clin Oncol. 2010;28(15 Suppl):abstractLBA7523
81. Takahashi T, Boku N, Murakami H. et al. Phase I and pharmacokinetic study of dacomitinib (PF-00299804), an oral irreversible, small molecule inhibitor of human epidermal growth factor receptor-1, -2, and -4 tyrosine kinases, in Japanese patients with advanced solid tumors. Invest New Drugs. 2012 Epub ahead of print
82. Ramalingam SS, Blackhall F, Krzakowski M. et al. Randomized Phase II Study of Dacomitinib (PF-00299804), an Irreversible Pan-Human Epidermal Growth Factor Receptor Inhibitor, Versus Erlotinib in Patients With Advanced Non-Small-Cell Lung Cancer. J ClinOncol. 2012;30(27):3337-3344
83. Tanner NT, Pastis NJ, Sherman C. et al. The role of molecular analyses in the era of personalized therapy for advanced NSCLC. Lung Cancer. 2012;76:131-137
84. Warth A, Penzel R, Brandt R. et al. Optimized algorithm for Sanger sequencing-based EGFR mutation analyses in NSCLC biopsies. Virchows Arch. 2012;460(4):407-14
85. Pareek C, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52(4):413-435
86. Su Z, Ning B, Fang H. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev Mol Diagn. 2011;11(3):333-43
87. Giaccone G, Wang Y. Strategies for overcoming resistance to EGFR family tyrosine kinase inhibitors. Cancer Treat Rev. 2011;37(6):456-464
88. Luping Lin, Trever G. Bivona. Mechanisms of resistance to epidermal growth factor receptor inhibitors and novel therapeutic strategies to overcome resistance in NSCLC patients. Chemother Res Pract. 2012 Epub ahead of print
89. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24-S31
90. Varella-Garcia M, Diebold J, Eberhard DA. et al. EGFR fluorescence in situ hybridisation assay: guidelines for application to non-small-cell lung cancer. J Clin Pathol. 2009;62(11):970-977
91. Pallis AG, Fennell DA, Szutowicz E. et al. Biomarkers of clinical benefit for anti-epidermal growth factor receptor agents in patients with non-small-cell lung cancer. Br J Cancer. 2011;105(1):1-8
92. Zhang X, Chang A. Molecular predictors of EGFR-TKI sensitivity in advanced non-small cell lung cancer. Int J Med Sci. 2008;5(4):209-217
93. Lim EH, Zhang SL, Yu K. et al. An alternative approach to determining therapeutic choices in advanced non-small cell lung carcinoma (NSCLC): maximizing the diagnostic procedure and the use of low-volume lung biopsies. J ThoracOnco. 2007;2:387-396
94. Shih JY, Gow CH, Yu CJ. et al. Epidermal growth factor receptor mutations in needle biopsy/aspiration samples predict response to gefitinib therapy and survival of patients with advanced non small cell lung cancer. Int J Cancer. 2006;118:963-969
95. Sunaga N, Tomizawa Y, Yanagitani N. et al. Phase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non-small cell lung cancer with EGFR mutations, irrespective of previous chemotherapy. Lung Cancer. 2007;56:383-389
96. Nakajima T, Yasufuku K, Sufuku M. et al. Assessment of epidermal growth factor receptor mutation by endobronchial ultrasound-guided transbronchial needle aspiration. Chest. 2007;132:597-602
97. Yoshida K, Yatabe Y, Park JY. et al. Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer. J Thorac Oncol. 2007;2:22-28
98. Garcia-Olivè I, Monsò E, Andreo F. et al. Endobronchial ultrasound-guidedtransbronchial needle aspiration for identifying EGFR mutations. Eur Respir J. 2010;35:391-5
99. Fassina A, Gazziero A, Zardo D. et al. Detection of EGFR and KRAS mutations ontrans-thoracic needle aspiration of lung nodules by high resolution melting analysis. J ClinPathol. 2009;62:1096-1102
100. Ellison G, Zhu G, Moulis A. et al. EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol. 2012;0:1-11
101. Kimura H, Kasahara K, Shibata K. et al. EGFR mutation of tumor and serum ingefitinib-treated patients with chemotherapy-naive non-small cell lung cancer. J ThoracOncol. 2006;1:260-267
102. Kim KS, Jeong JY, Kim YC. et al. Predictors of the response to gefitinib in refractorynon-small cell lung cancer. Clin Cancer Res. 2005;11:2244-2251
103. Mack PC, Holland WS, Burich RA. et al. EGFR mutations detected in plasma areassociated with patient outcomes in erlotinib plus docetaxel-treated non-small celllung cancer. J ThoracOncol. 2009;4:1466-1472
104. Mattsson JS, Imgenberg-Kreuz J, Edlund K. et al. Consistent mutation status within histologically heterogeneous lung cancer lesions. Histopathology. 2012;61(4):744-748
105. Chen ZY, Zhong WZ, Zhang XC. et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist. 2012;17(7):978-85
106. Sun L, Zhang Q, Luan H. et al. Comparison of KRAS and EGFR gene status between primary non-small cell lung cancer and local lymph node metastases: implications for clinical practice. J ExpClin Cancer Res. 2011;30(1):30
107. Jakobsen JN, Sorensen JB. Intratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancer. Cancer ChemotherPharmacol. 2012;69(2):289-99
Author contact
![]() Corresponding author: Cafarotti Stefano, MD. Thoracic Surgery EOC Unit - San Giovanni Hospital, Bellinzona, Switzerland. cafarottistefanocom. Tel 0039 3207660702 / 0041 (0) 787949238 Fax 0039 0630156329.
Corresponding author: Cafarotti Stefano, MD. Thoracic Surgery EOC Unit - San Giovanni Hospital, Bellinzona, Switzerland. cafarottistefanocom. Tel 0039 3207660702 / 0041 (0) 787949238 Fax 0039 0630156329.
Received 2012-5-17
Accepted 2012-12-28
Published 2013-2-11