ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2008; 5(2):92-99. doi:10.7150/ijms.5.92 This issue Cite
Research Paper
Vgf is a novel biomarker associated with muscle weakness in amyotrophic lateral sclerosis (ALS), with a potential role in disease pathogenesis
Zhong Zhao1,2, Dale J. Lange1,3, Lap Ho1,2, Sara Bonini1,2, Belinda Shao1,2, Stephen R. Salton4,5, Sunil Thomas1,2, Giulio Maria Pasinetti1,2,4,5 ![]()
1. James J. Peters Veterans Affairs Medical Center, Bronx, NY 10468
2. Departments of Psychiatry, Mount Sinai School of Medicine, New York, NY-10029
3. Departments of Neurology, Mount Sinai School of Medicine, New York, NY-10029
4. Departments of Neuroscience, Mount Sinai School of Medicine, New York, NY-10029
5. Departments of Geriatrics, Mount Sinai School of Medicine, New York, NY-10029
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects nerve cells in the brain and the spinal cord. Previous proteomic evidence revealed that the content of certain peptide fragments including Vgf-derived peptide aa 398-411 (Vgf398-411) of the precursor Vgf protein in the cerebral spinal fluid (CSF) correctly identified patients with ALS from normal and disease controls. Using quantitative ELISA immunoassay we found that the CSF levels of Vgf decreases with muscle weakness in patients with ALS. In SOD1 G93A transgenic mice, loss of full-length Vgf content in CSF, serum and in SMI-32 immunopositive spinal cord motor neurons is noted in asymptomatic animals (approximately 75 days old) and continues to show a progressive decline as animals weaken. In vitro studies show that viral-mediated exogenous Vgf expression in primary mixed spinal cord neuron cultures attenuates excitotoxic injury. Thus, while Vgf may be a reliable biomarker of progression of muscle weakness in patients with ALS, restoration of Vgf expression in spinal cord motor neurons may therapeutically rescue spinal cord motorneurons against excitotoxic injury.
Keywords: VGF, ALS, biomarker, excitotoxicity, SOD-1 mice, disease progression
Introduction
Vgf nerve growth factor inducible peptide, a non acronymic peptide is a member of the chromogranin/secretogranin family of proteins. These proteins are stored in large dense core vesicles (LDCVs) located in neuroendocrine, endocrine and neuronal cells. LDCVs are released in response to depolarization and other stimuli. Vgf is widely expressed in the central nervous system including spinal cord motorneurons. We previously reported that decreased CSF levels of certain peptide fragments including Vgf398-411 correctly identify patients with ALS from normal and disease controls. [1] The biological role of Vgf is incompletely understood, [2-13] although recent studies demonstrate significant endocrine, metabolic and anti-depressant effects of Vgf-derived peptides. [14-17]
The present study suggests that Vgf may be a useful biomarker to monitor ALS onset and clinical progression and that therapeutic preservation of Vgf might neuroprotect spinal cord motorneurons against excitotoxic injury in ALS.
Methods
Human subjects
CSF from normal subjects (n=21) and ALS patients (n=17) were used for ELISA. ALS patients were classified as having either definite or probable ALS according to the WFN El-Escorial diagnostic criteria. [18] ALS patients were classified according to number of segments with clinical weakness, from a total of 3 segments of the central nervous system (cranial, cervical, and lumbar). Clinical weakness identified only in one segment occurred in 10 patients; weakness in two segments was identified in 7 patients. The total score on manual muscle testing (MMT) measured severity of muscle weakness. Five muscle groups in each of the four limbs were examined and graded according to the standard Medical Research Council (MRC) criteria, on a scale from 0 (no movement) to 5 (full strength against maximal resistance). The total possible normal score on this examination is 100. All CSF samples used were derived from comparable fractions (e.g. 20–25 ml), to limit variability from rostro-caudal concentration gradients. Following collection, samples were gently mixed, divided into aliquots, and immediately frozen in dry-ice and stored at –80° C. Written informed consent, approved by the Mount Sinai School of Medicine Institutional Review Board (IRB) on November 3, 2004, was obtained from all subjects.
ELISA assays
hVgf ELISA, microtiter plates were coated with 2-mg/mL goat polyclonal anti-Vgf antibody (R15) (Santa Cruz Biotech, Santa Cruz, CA), which recognizes the C-terminal epitope of Vgf. Unoccupied binding sites on the plates were blocked by incubation with casein. Samples and standards were applied in duplicate and incubated overnight at 4°C. Following the Vgf capture phase, the plates were reacted with rabbit anti-Vgf antibody (#9130 against Vgf78-340) [19] followed by incubation with a reporter antibody (HRP–conjugated anti–rabbit IgG, Santa Cruz Biotech, CA). The assay was developed using a stabilized HRP substrate. All samples were analyzed in the linear range of the ELISA using over-expressed human Vgf as a standard.
Assessment of motor function
Mutant G93A SOD-1 transgenic mice were tested on the accelerating Rotarod (7650 Ugo Basile Biol. Res. App., Comerio, Italy) as described previously. [20-21] Mice were tested 3 times a week beginning at ~ 70 days, until the transgenic mice could no longer perform the tests. Before testing, mice underwent a one-week training period wherein they were introduced to the apparatus and handled by the operator daily. Testing was conducted during the last 4 hours of the day portion of the light cycle in an environment with minimal stimuli (noise, movement, changes in light or temperature) for a maximum time maintained on the rod by each mouse of 240 seconds.
Western blot and protein expression analysis
Frozen brain and spinal cord samples were first pulverized on dry ice, homogenized in cell lysis buffer (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1mM phenylmethyl sulphonyl fluoride), and sonicated for 2 min at 4ºC. The lysates were centrifuged at 2,500 x g for 15 min at 4ºC to remove nuclei and cell debris. Samples were then separated (50-100 μg protein loaded per lane) on 12% SDS-PAGE, transferred to a PVDF membrane (BioRad), and detected with rabbit anti-Vgf antibody (Ab5901, 1:1000).[22] On the same membrane, β-tubulin (rat anti-β tubulin, 1:5000, Sigma, St. Louis, MO) was used to control sample loading and to normalize Vgf immunoreactivity.
Immunocytochemistry
For semiquantitative assessment of Vgf in spinal cord motorneurons, immunohistochemistry was performed as previously reported [21] with minor modifications. Briefly, spinal cord tissue sections were treated with an antibody against Vgf (rabbit anti rat monoclonal D20, 1:1000, Santa Cruz, CA) or against SMI-32 (rabbit polyclonal, 1:200 dilution; Santa Cruz Biotechnology, Inc.) labeled with Zenon Alexa Fluor 594 (mouse IgG labeling kit; Molecular Probes Inc.) at 25°C for 1 h. Furthermore, the sections were treated with the monoclonal antibody against Vgf (rat monoclonal, 1:100 dilution; Santa Cruz Biotechnology, Inc.) labeled with Zenon Alexa Fluor 488 (mouse IgG labeling kit; Molecular Probes Inc.) at 25°C for 1 h. The fluorescence emitted was observed through each appropriate filter on a fluorescence microscope (BX51; Olympus) and digitally photographed using a cooled charge-coupled-device camera (model VB-6000/6010; Keyence Co.). In control studies run in parallel, tissue sections were also stained with anti- glial fibrillary acidic protein (GFAP), a glial marker, or and anti NeuN, a neuronal marker, as previously described.[21]
Stereology of SMI-32 immunopositive neurons
For stereological assessment of SMI-32 (a non-phosphorylated neurofilament epitope) immunoreactive spinal cord motorneurons, 10 serial coronal sections (12 μm thick) were cut 350 μm apart through the lumbar (L3 to L5) spinal cord of each animal. The sections were mounted onto positively charged glass slides (Superfrost Plus, Fisher Scientific) and immunostained using a commercially available rabbit anti-rat SMI-32 antibody (D20, 1:1000, Santa Cruz, CA). SMI-32 immunopositive neurons were counted from digitised images (200X) within the ventral horns under fluoresce microscopy. These counts were within a homogenous structure, making the tenets of stereology valid. SMI-32-stained neurons were counted using the Neurolucida system at a magnification of 250X in both ventral horn areas from six L3-L5 tissue sections of the spinal cord of each mouse. All SMI-32 immunoreactive neurons were counted from within the ventral horn below a lateral line across the spinal cord from the central canal. Correction for tissue section thickness was made in all specimens.
For stereological analysis, 10 serial coronal sections (12μm thick) were cut 350μm apart through the lumbar (L3 to L5) spinal cord of each animal (WT controls; n=6).
Semiquantitative Vgf quantification in spinal cord motorneurons
The immunostaining densities were digitized with a high-resolution fluorescence charge-coupled-device camera (Sony, Tokyo, Japan) and semi-quantified using Bioquant computer-assisted densitometry (Biometrics, Nashville, TN). Fluorescence camera aperture and focus were adjusted to provide an optimal image. The overall illumination was also adjusted so that the distribution of relative values fell within the limits of the system avoiding a floor or ceiling effect. Once established, the setting remained constant for all the images acquired for all the ICC experiments. Therefore, when all the parameters were fixed, only tissue staining intensities influenced the measured values. Average value density measurements from individual Vgf immunoreactive dorsal spinal cord neurons, reflecting immunostaining intensity, were made on digitized images by delimiting the cellular area of interest free hand, using predetermined criteria to define the region of interest. The immune intensity of the cellular Vgf encompassing the L3-L5 regions of spinal cord were semi-quantified from approximately 6 to 8 frames per spinal cord tissue sections; about 3 to 10 neurons per frame were randomly quantified. The technician who performed these measurements had no knowledge of the study groups. To normalize any unevenness in lighting across the field of view; background values were determined over the white matter area of each individual tissue section and automatically subtracted from the values over unstained spinal cord motorneurons. The optical density of cellular Vgf immunostaining in spinal cord dorsal neurons was expressed using arbitrary units.
Mouse Vgf radio immuno assay (RIA)
C-terminal specific Vgf antibody (ab5901) was used in RIA analysis as previously described [22] to detect full-length Vgf and processed Vgf peptides containing the C-terminus. AQEE30 peptide was radiolabelled with I125 at ~2000 Ci/mmol specificity by GE-Healthcare (Woburn, MA). Briefly, samples or standard AQEE30 peptide, from 30-3000 fmol, were incubated with anti-Vgf (AQEE30) antibody (1:3000 dilution) in 200 µl RIA buffer (50mM Tris-Cl, 0.1% BSA, 0.1% Triton-X100, 0.1% Gelatin, 0.02% Sodium Azide) at 4°C overnight. After adding 100 µl of I125-AQEE30 tracer (10,000 cpm) at 4°C overnight, the antibody complex was precipitated with 100 µl of goat anti rabbit IgG and 10 µl of normal rabbit serum (Peninsula Laboratories Inc., San Carlos, CA) dissolved in RIA buffer. After incubating at room temperature for 1.5 hr, the reactions were stopped by addition of 250 µl ice-cold termination buffer (50mM Tris-HCl, 0.1% Triton-X100, 0.02% sodium azide). The supernatants were aspirated after centrifugation at 3700 x g for 20 min. Vgf-specific radioactivity was quantified using a CobraII Auto γ Counter (PerkinElmer, Wellesley, MA).
Adeno-Vgf viral constructs
The replication-defective recombinant-adeno-expression virus was generated using the Adeno-X expression system following the manufacturer's procedure (Clontech, CA). Briefly, mouse Vgf cDNA (Salton, unpublished data) was isolated via Xba I-Apa I restriction cleavage, and cloned into the NheI-ApaI sites of a pShuttle vector to generate the expression cassette under regulation of the cytomegalovirus (CMV) promoter. The cDNA constructs were sequence-confirmed, and the expression cassette was then transferred from the pShuttle construct into purified Adeno-X viral DNA via I-CeuI and PI-SceI sites.
The recombinant viral cDNA construct was confirmed by nucleotide sequencing, and the recombinant virus was packaged by infecting the PacI linearized recombinant viral DNA into human embryonic kidney (HEK)-293 cells (Clontech, CA). The resulting recombinant virus was further propagated in HEK 293 cells, and a viral titer was determined by tissue culture infectious dose 50 (TCID50). [23] A control Lac-Z recombinant adenovirus expressing the β-galactosidase gene product was prepared using an identical strategy. HEK 293 cells were cultured in DMEM medium supplemented with 10% fetal calf serum (FBS) and 100 U/ml penicillin-streptomycin (Gibco, NY).
Spinal cord neuron cultures
Mixed spinal cord cultures were prepared according to Zhao et al.[21]. Briefly, spinal cord cultures were prepared from E14 embryos dissected from pregnant wild type females that had been mated with SOD1-G93A transgenic males. Each spinal tube was dissected, removed from the meninges, and incubated for 10 min in 0.25% trypsin/EDTA at 37°C and then dissociated by gentle trituration with a fire-polished Pasteur pipette. The cell suspension was plated in D-MEM/F12 supplemented with 10% FBS on a poly D lysine-coated 96 well plate at a density of 105 cells/well. After 30 min, the medium was replaced with Neurobasal media supplemented with 2% B-27, 0.5 mM glutamine, and 1% penicillin/streptomycin. Cultures were maintained under standard conditions as previously reported [21].
Excitotoxicity studies in vitro
In viral expression studies, 5 day-old cultures were replaced with fresh Neurobasal medium containing Adeno-LacZ or Adeno-Vgf constructs, at a multiplicity of infection (MOI) of 5, and culture medium was replaced again 3 days thereafter. For excitotoxicity studies ~ 8-day-old spinal cord cultures were challenged with the glutamate receptor agonists AMPA (5 µM) and NMDA (20 µM) for 48 hours. Neurotoxicty was assessed by LDH assay kit according the manufacturer's instructions (Promega Corp Madison, WI), or using an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay.[24]
Statistical analysis
Statistical analyses were performed using SigmaStat (version 3.0, SPSS Inc., Chicago, IL). Independently measured t-tests were used to compare endpoints between control and experimental groups in either clinical or preclinical studies. ANOVA tests among groups were followed, when significant, by the Student-Newman-Keuls multiple comparison tests. The Student-Newman-Keuls test was chosen for post-hoc multiple comparisons due to its generally higher sensitivity compared to the Bonferroni test. In all tests, results with probability values less than 0.05 were considered statistically significant. Presented data are shown as mean ± SEM, unless otherwise noted. The receiver operating characteristic (ROC) curve was used to relate “sensitivity” and “specificity,” or sensitivity at a given specificity for providing cut-off values, as previously described [1]. The null hypothesis was rejected at the 0.05 level in all analyses.
Results
CSF Levels of Vgf correctly diagnose ALS and associates with clinical severity
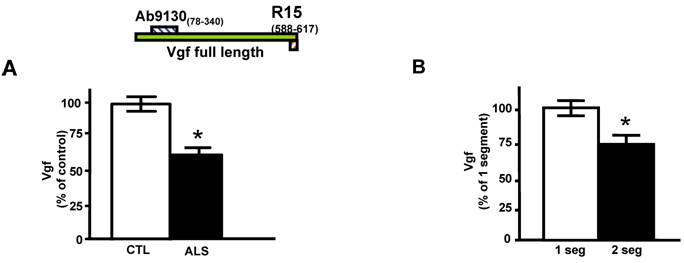
Quantitative ELISA assay revealed that the decreased CSF levels of total full-length Vgf (P<0.05), correctly diagnosed ALS patients with 77% sensitivity and 87% specificity based on receiving operating characteristic (ROC) analysis (Figure 1A). Vgf CSF content decreased as a function of progression of muscle weakness characterized by an increasing number of affected muscle (segments) assessed by manual muscle testing (P<0.05) (Figure 1B).
Full length Vgf content in CSF in ALS. In A, full-length Vgf was assessed by quantitative ELISA assays; in B, Vgf content decreased as a function of progression of muscle weakness assessed by manual muscle testing revealing an increased number of affected muscle (segments). Quantitative muscle testing was based on the MRC clinical grading system, out of a total of 100 possible points. ROC analysis was carried out to determine the sensitivity and specificity of Vgf in dissecting control vs. ALS subjects. Values are expressed as percent of control level (mean ± SEM; * 2-tailed t-test, p<0.05). Inset, Vgf protein sequence used to raise Vgf antibodies for ELISA assays (see Materials and Methods for more information).
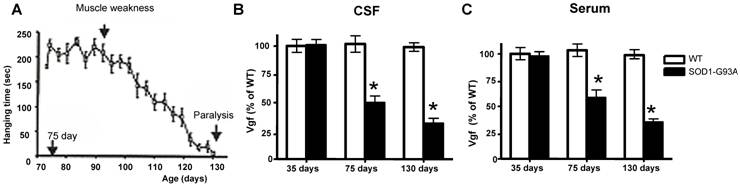
Decreased Vgf content In CSF and serum precedes onset of ALS-type muscle weakness assessed by rotarod-assays
In our laboratory setting, G93A mutant SOD-1ALS mice develop muscle weakness by ~90 days of age (Figure 2A). The severity of motor impairment progresses to paralysis by ~130 days of age, followed by sacrifice.[1] No detectable change in Vgf content in CSF and serum of G93A SOD-1 ALS mice was found in ~35 days old G93A SOD-1 ALS mice, relative to age-, gender-, and strain-matched wild-type littermates (Figure 2B,C).
Reduction in Vgf content in the CSF (F1,7793=4.913, P=0.0288 for age, F7,23660=2.131, P=0.0466 for Vgf content) and in the serum (F1,19840=5.4573, P=0.0345 for age, F7,34510 =3.945, P=0.0487 for Vgf content) in ~75 days old G93A SOD-1 mice was found to precede the onset of muscle weakness assessed by rotarod assay that normally occurs at ~90 days, relative to age-, gender-, and strain-matched wild-type littermates.
Serum and CSF content of Vgf continued to decrease up to ~130 days of age when G93A-SOD1 ALS mice are characterized by near complete paralysis, relative to gender-matched WT littermates (Figure 2B, C).
Vgf immunoreactive material in the lumbar (L3 to L5) spinal cord colocalizes with SMI-32 immunopositive motorneurons and decreases as a function of age progression in SOD-1 ALS mice
In the lumbar L3 to L5 region of spinal cord the distribution of Vgf immunoreactive material colocalized with SMI-32 immunoreactive spinal cord neurons (Figure 3A, panels 2-4). No detectable Vgf immunopositive signal was found to co-localize with NeuN immunoreactive neurons in the same spinal cord region (Figure 3B, panels 2-4), nor was there Vgf immunoreactive signal that co-localized with GFAP immunopositive astrocytes (data not shown).
Decreased Vgf content in the CSF and serum precedes ALS-type motor impairment assessed by rotarod assay. In A, ALS-type muscle weakness in mutant G93A SOD-1 as a function of clinical progression (age). In B,C, decreased Vgf levels in CSF and in serum respectively precedes ALS-type muscle weakness in ~90 day-old symptomatic mutant G93A-SOD-1 mice and continue to decline as a function of progression of ALS-type clinical disease. Values are expressed as mean ± SEM; n=4-5 per group; 2-way ANOVA. No detectable muscle weakness was found in age-gender matched WT controls at any time examined (not shown).
Vgf immunoreactive material in the lumbar spinal cord co-localizes with SMI-32 immunopositive motorneurons and decreases as a function of age progression of SOD-1 ALS mice. In A, Vgf immunoreactive material is selectively localized within the nuclear region of SMI-32 immunoreactive spinal cord neurons. In B, no detectable Vgf co-localization with NeuN immunoreactive neurons. In C, Vgf immunoreactive material in spinal cord motorneurons as a function of age. In D, SMI-32 spinal cord motorneurons in the L3-L5 region of spinal cord in ~130 days old mutant G93A-SOD1 ALS mice. Values are expressed as mean ± SEM; n=4-5 per group; In C,*2-way ANOVA; in D, *p<0.05 by 2-tailed t-test
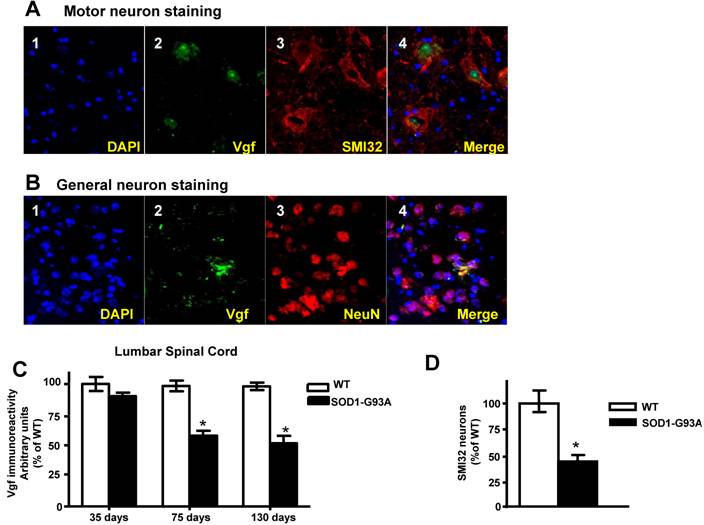
Survey of Vgf content assessed immunocytochemically revealed that Vgf immunoreactive material in spinal cord motorneurons is already decreased in ~75 day old asymptomatic SOD-1 G93A-SOD1 ALS mice and continue to decrease as a function of progression of ALS-type muscle weakness up to ~130 days of age (Figure 3C)(F1,19840=14.28, P=0.0003 for age, F7,34510 =3.549, P=0.0018 for Vgf content), relative to age-, gender-, and strain-matched wild-type littermates.
The loss of Vgf immmunoreactive signal in SMI-32 spinal cord motorneurons in the L3-L5 region of spinal cord in ~130 days old mutant G93A-SOD1 ALS mice overlaps quantitatively with the loss in SMI-32 immunoreactive motorneurons assessed stereologically in the same tissue sections (Figure 3D), relative to age-, gender-, and strain-matched wild-type littermates.
Exogenous adenoviral Vgf expression protects G93A SOD1 mixed spinal cord neurons against excitotoxic injury
Total full-length mouse (m)vgf protein was expressed in ~8 days old primary mix mutant G93A SOD-1 spinal cord neuron cultures derived from E14 mouse embryos by infection with an adenoviral Vgf (Ad)-Vgf at 5 MOI.
Under this experimental condition, Ad-mVgf expression significantly increased intracellular Vgf and secreted full length Vgf protein (85 kDa) in the conditioned medium, relative to parallel Lac-Z infected cultures (5 MOI) (Figure 4, and Figure 4 inset (not shown)), as assessed by western blot 48 hrs after infection. Next we explored the influence of exogenous Vgf expression in response to excitotoxic neuronal injury.
Exogenous adenoviral mVgf expression attenuates excitotoxicity in mixed cultures of spinal cord neurons. Neuron cultures were infected with Adeno (Ad)-Vgf or Ad-LacZ viral constructs at 5 MOI for 72 hrs, and then treated with AMPA (5 µM), NMDA (20 µM) for 48 hrs while cell viability was assessed by LDH. Values are expressed as mean ± SEM as % of control (CTL) group; n=3 independent cultures per group; *P<0.05, **P<0.01; Ad-Vgf treatment vs. Ad-Lac-Z infected controls.
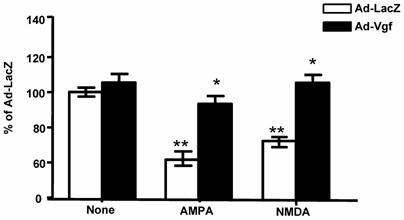
We found treatment of control Lac-Z infected (5 MOI) primary mixed spinal cord neurons with glutamate receptor agonist AMPA (5 µM) or NMDA (20 µM), results in significant 30-40% loss in cell viability relative to untreated Lac-Z infected cultures (P<0.01; P<0.05, respectively), as assessed by LDH cell viability assay, 48 hr after treatment (Figure 4).
We found that preventative expression of Ad-Vgf in primary mix mutant G93A SOD-1 spinal cord neuron cultures for 48 hrs resulted in significant protection against AMPA or NMDA mediated excitotoxic injury, as assessed by a neuronal viability assay 48 hr following drug treatment (Figure 4).
Discussion
Our study demonstrates that reduced levels of full-length Vgf content in the CSF of ALS patients may be a useful measure of disease progression, as assessed by scores of muscle strength. Consistently, in experimental studies we found that Vgf content in the CSF, serum and spinal cord motorneurons is a specific predictor of onset and progression of clinical motor impairment. Thus, Vgf could provide a biological index to predict and monitor the development of motor impairment in ALS before onset of clinical disease. Our study supports the hypothesis that restoring Vgf expression in spinal cord motor neurons could therapeutically benefit clinical ALS.
The mechanism(s) that underlie the relationship between mutations in the SOD-1 gene and the pathogenesis of sporadic ALS are uncertain. However, experimental evidence suggests that mitochondrial dysfunction/degeneration resulting in abnormal energy metabolism are important factors in the evolution of the disease in several SOD-1 mutant mouse models of ALS, including the G93A-SOD1 ALS mice used in this study. [25-30]
Evidence suggests that both mutant SOD-1 ALS [3] and Vgf knockout [5,31,32] mice similarly display abnormal energy expenditure features supporting the hypothesis that reduced Vgf levels in an ALS model mice could contribute to the lean, hypermetabolic state that eventually could negatively influence clinical disease in ALS. Thus our study for the first time tentatively supports the hypothesis that a mechanism thorough which abnormal metabolism may ultimately influence ALS pathogenesis is through mechanisms involving reduced expression of Vgf. This evidence is of high interest to this study especially in view of previous reports suggesting that sporadic ALS patients can be hypermetabolic with decreased fat mass,[25] a phenotype resembling that found in mutant G93A-SOD1 ALS mice.[3]
We found that exogenous viral expression of Vgf neuroprotects primary mixed spinal cord neurons against excitotoxicity, which is a pathogenic feature in ALS.[20] This observation tentatively suggests that reduced levels of Vgf expression in spinal cord motorneurons in ALS SOD-1 ALS mice could mechanistically promote neurodegeneration by unleashing NMDA and AMPA excitotoxic injury. Thus a mechanism by which abnormal energy metabolism may have an influence on clinical ALS is through depletion of Vgf neuroprotection against spinal cord motorneuron excitotoxic injury.
Collectively our study suggests that reduced Vgf content in the serum, in the CSF, and in spinal cord motorneurons may be a biological diagnostic index for ALS. It remains to be directly tested whether restoring Vgf expression in spinal cord motor neurons therapeutically rescues the ALS phenotype.
Acknowledgements
Supported by ALS grant from the Department of Veterans Affairs, NCCAM 5R21 AT002602-02 and NCCAM 1R21 AT003632-01A1 to GMP and NARSAD and DK071308 to SRS.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Pasinetti GM, Ungar LH, Lange DJ, Yemul S, Deng H, Yuan X, Brown RH, Cudkowicz ME, Newhall K, Peskind E, Marcus S, Ho L. Identification of potential CSF biomarkers in ALS. Neurology. 2006;66(8):1218-1222
2. Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, Black IB. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of Vgf in hippocampal synaptic plasticity. J Neurosci. 2003;23(34):10800-10808
3. Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101(30):11159-11164
4. Garcia AL, Han SK, Janssen WG, Khaing ZZ, Ito T, Glucksman MJ, Benson DL, Salton SR. (2005) A prohormone convertase cleavage site within a predicted alpha-helix mediates sorting of the neuronal and endocrine polypeptide Vgf into the regulated secretory pathway. J Biol Chem. 2005;280(50):41595-41608
5. Hahm S, Mizuno TM, Wu TJ, Wisor JP, Priest CA, Kozak CA, Boozer CN, Peng B, McEvoy RC, Good P, Kelley KA, Takahashi JS, Pintar JE, Roberts JL, Mobbs CV, Salton SR. Targeted deletion of the Vgf gene indicates that the encoded secretory peptide precursor plays a novel role in the regulation of energy balance. Neuron. 1999;23(3):537-548
6. Hahm S, Fekete C, Mizuno TM, Windsor J, Yan H, Boozer CN, Lee C, Elmquist JK, Lechan RM, Mobbs CV, Salton SR. VGF is required for obesity induced by diet, gold thioglucose treatment, and agouti and is differentially regulated in pro-opiomelanocortin- and neuropeptide Y-containing arcuate neurons in response to fasting. J Neurosci. 2002;22(16):6929-6938
7. Hawley RJ, Scheibe RJ, Wagner JA. NGF induces the expression of the Vgf gene through a cAMP response element. J Neurosci. 1992;12(7):2573-2581
8. Levi A, Eldridge JD, Paterson BM. Molecular cloning of a gene sequence regulated by nerve growth factor. Science. 1985;229(4711):393-395
9. Salton SR, Fischberg DJ, Dong KW. Structure of the gene encoding Vgf, a nervous system-specific mRNA that is rapidly and selectively induced by nerve growth factor in PC12 cells. Mol Cell Biol. 1991;11(5):2335-2349
10. Snyder SE, Salton SR. Expression of Vgf mRNA in the adult rat central nervous system. J Comp Neurol. 1998;394(1):91-105
11. Snyder SE, Pintar JE, Salton SR. Developmental expression of Vgf mRNA in the prenatal and postnatal rat. J Comp Neurol. 1998;394(1):64-90
12. van den Pol AN, Decavel C, Levi A, Paterson B. Hypothalamic expression of a novel gene product, Vgf: immunocytochemical analysis. J Neurosci. 1989;9(12):4122-4137
13. van den Pol AN, Bina K, Decavel C, Ghosh P. Vgf expression in the brain. J Comp Neurol. 1994;347(3):455-469
14. Jethwa PH, Warner A, Nilaweera KN, Brameld JM, Keyte JW, Carter WG, Bolton N, Bruggraber M, Morgan PJ, Barrett P, Ebling FJ. VGF-derived peptide, TLQP-21, regulates food intake and body weight in Siberian hamsters. Endocrinology. 2007;148(8):4044-4055
15. Bartolomucci A, La Corte G, Possenti R, Locatelli V, Rigamonti AE, Torsello A, Bresciani E, Bulgarelli I, Rizzi R, Pavone F, D'Amato FR, Severini C, Mignogna G, Giorgi A, Schinina ME, Elia G, Brancia C, Ferri GL, Conti R, Ciani B, Pascucci T, Dell'Omo G, Muller EE, Levi A, Moles A. TLQP-21, a VGF-derived peptide, increases energy expenditure and prevents the early phase of diet-induced obesity. Proc Natl Acad Sci USA. 2006;103(39):14584-14589
16. Yamaguchi H, Sasaki K, Satomi Y, Shimbara T, Kageyama H, Mondal MS, Toshinai K, Date Y, Gonzalez LJ, Shioda S, Takao T, Nakazato M, Minamino N. Peptidomic identification and biological validation of neuroendocrine regulatory peptide-1 and -2. J Biol Chem. 2007;282(36):26354-26360
17. Hunsberger JG, Newton SS, Bennett AH, Duman CH, Russell DS, Salton SR, Duman RS. Antidepressant actions of the exercise-regulated gene VGF. Nat Med. 2007;13(12):1476-1482
18. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299
19. Salton SR, Volonte C, D'Arcangelo G. Stimulation of vgf gene expression by NGF is mediated through multiple signal transduction pathways involving protein phosphorylation. FEBS Lett. 1995;360(2):106-110
20. Pompl PN, Ho L, Bianchi M, McManus T, Qin W, Pasinetti GM. A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2003;17(6):725-727
21. Zhao Z, Lange DJ, Voustianiouk A, Macgrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti GM. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006;7:29
22. Chakraborty TR, Tkalych O, Nanno D, Garcia AL, Devi LA, Salton SR. Quantification of Vgf- and pro-SAAS-derived peptides in endocrine tissues and the brain, and their regulation by diet and cold stress. Brain Res. 2006;1089(1):21-32
23. Nyberg-Hoffman C, Shabram P, Li W, Giroux D, Aguilar-Cordova E. Sensitivity and reproducibility in adenoviral infectious titer determination. Nat Med. 1997;3(7):808-811
24. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1-2):55-63
25. Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrere B, Couratier P. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr. 2001;74(3):328-334
26. Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18(9):3241-3250
27. Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277(33):29626-29633
28. Menzies FM, Ince PG, Shaw PJ. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem Int. 2002;40(6):543-551
29. Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43(1):19-30
30. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14(6):1105-1116
31. Levi A, Ferri GL, Watson E, Possenti R, Salton SR. Processing, distribution, and function of Vgf, a neuronal and endocrine peptide precursor. Cell Mol Neurobiol. 2004;24(4):517-533
32. Salton SR, Ferri GL, Hahm S, Snyder SE, Wilson AJ, Possenti R, Levi A. Vgf: a novel role for this neuronal and neuroendocrine polypeptide in the regulation of energy balance. Front Neuroendocrinol. 2000;21(3):199-219
Author contact
![]() Correspondence to: Dr. Giulio Maria Pasinetti, Mount Sinai School of Medicine, Department of Psychiatry, One Gustave L., Levy Place, Box 1668, New York, NY-10029. Email: giulio.pasinettiedu
Correspondence to: Dr. Giulio Maria Pasinetti, Mount Sinai School of Medicine, Department of Psychiatry, One Gustave L., Levy Place, Box 1668, New York, NY-10029. Email: giulio.pasinettiedu
Received 2008-2-25
Accepted 2008-4-12
Published 2008-4-15