ISSN: 1449-1907International Journal of Medical Sciences
- Current issue
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Volume 18; 2021
- Volume 17; 2020
- Archive
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. Introduction
2. The attachment of HBV
3. The cell receptor for HBV...
4. The HBV fusion and proteolysis
5. Internalization of HBV...
6. Over-expression of Serine...
7. Systems used for the study of...
Acknowledgements
References
Figures
Author biography

Int J Med Sci 2004; 1(1):21-33. doi:10.7150/ijms.1.21 This issue Cite
Study of the early steps of the Hepatitis B Virus life cycle
Xuanyong Lu
, Timothy Block
![]()
Jefferson Center for Biomedical Research and Agricultural Medicine, Department of Biochemistry and Molecular Pharmacology, Thomas Jefferson University, Philadelphia, USA
Abstract
Hepatitis B virus (HBV) is a human pathogen, causing the serious liver disease. Despite considerable advances in the understanding of the natural history of HBV disease, most of the early steps in the virus life cycle remain unclear. Virus attachment to permissive cells, fusion and penetration through cell membranes and subsequent genome release, are largely a mystery. Current knowledge on the early steps of HBV life cycle has mostly come from molecular cloning, expression of individual genes and studies of the infection of duck hepatitis B virus (DHBV) with duck primary duck hepatocytes. However, considering of the difference of the surface protein of HBV and DHBV both in the composition and sequence, the degree to which information from DHBV applies to human HBV attachment and entry may be limited. A major obstacle to the study HBV infection is the lack of a reliable and sensitive in vitro infection system. We have found that the digestion of HBV and woodchuck hepatitis virus (WHBV) by protease V8 led to the infection of HepG2 cell, a cell line generally is refractory for their infection [Lu et al. J Virol. 1996. 70. 2277-2285. Lu et al. Virus Research. 2001. 73(1): 27-4]. Further studies showed that a serine protease inhibitor Kazal (SPIK) was over expressed in the HepG2 cells. Therefore, it is possible that to silence the over expressed SPIK and thus to reinstate the activity of indispensable cellular proteases can result in the restoration of the susceptibility of HepG2 cells for HBV infection. The establishing a stable cell line for study of the early steps of HBV life cycle by silencing of SPIK is discussed.
Keywords: Hepatitis B virus, Fusion, Serine protease inhibitor, Receptor, Attachment.
1. Introduction
The Hepatitis B virus (HBV), which belongs to the hepadnavirus family, is a small circular DNA virus containing a nucleocapsid and an envelope. HBV nucleocapsid contains a relatively small and incompletely double stranded 3.2 Kb DNA genome, viral polymerase and core protein. Its envelope is composed of viral surface proteins enclosed by a lipid membrane from host cells [1, 2]. In the serum of infected patients, there are both mature virion with viral DNA and subviral particles without viral DNA [3, 4]. Sub viral particles are overwhelmingly in excess to infectious particles, which is the majority of the two types [3, 4]
The life cycle of HBV is believed to begin when the virus attaches to the host cell membrane via its envelope proteins. Then, the viral membrane fuses with the cell membrane and the viral genome is released into the cells [5, 6] After the viral genome reaches the nucleus, the viral polymerase converts the partial double stranded DNA (dsDNA) genome into covalently closed circle DNA (cccDNA). The cccDNA is believed to be the template for further propagation of pre-genomic RNA, which directs the synthesis of viral DNA and mRNA that encode all the viral proteins [2, 7, 8] HBV core particles are assembled in the cytosol following the encapsidation of pre-genomic RNA, which is then degraded during the reverse transcription of pre-genomic RNA into a complementary strand of DNA [9]. HBV surface proteins are initially synthesized and polymerized in the rough endoplasmic reticulum (RER). These proteins are transported to the post ER and pre-Golgi compartments where budding of the nucleocapsid follows [10]. The assembled HBV virion and sub-viral particles are transported to the Golgi for further modification of its glycans in the surface proteins, and then are secreted out of the host cell to finish the life cycle. HBV replication and viral protein synthesis in the infected cells are fairly well elucidated. However, the early steps of HBV infection including of the penetration of virus and release of its genome into host cells is uncertain.
In general, the early step of virus infection in which the virus enters the cell can be divided into three stages: attachment, fusion, and entry. Enveloped viruses usually entry by an attachment to the host cells, which usually is via the interaction of viral surface protein with the specific receptor on the cell membrane [11]. However, the attachment itself does not necessarily result in viral entry. The fusion of a viral envelope and cell membrane and the following viral genome release finally trigger the viral infection [12, 13]. The viral fusion occurs by one of at least two known mechanisms. The first requires a fusion of the viral envelope with the plasma membrane, leading to the release of the viral nucleocapsid. HIV is an example of a virus that uses this type of mechanism to enter [12]. In the second mechanism, an endosomal compartment first takes up the attached virus. Later, the viral genome is released from this endosomal compartment into the cytoplasm by a process that may or may not depend upon a lowering of the pH to activate a virus-mediated fusogenic activity. In some virus/cell systems, such as those involving influenza virus [14] or paramyxoviruses [11, 13] exogenously adding protease generates infective virions from otherwise non-infective ones. The reason is that the proteolysis exposes the viral fusion sequence. The un-coating and the genome release occur instantly after virus fusion. The following transport of the viral genome to nucleus and the start of the virus replication finally complete the early steps of virus life cycle.
2. The attachment of HBV
A major limitation to study HBV early steps of life cycle is lack of an in vitro infection system. Although HBV is considered highly efficient in establishing infection in people following parenteral exposure, tissue culture cells are curiously refractory for HBV infection [5, 15, 16, 17]. Although there is a growing body of data, recently most of the published information about the early stages of hepadnavirus infection is derived from duck hepatitis B virus (DHBV), since robust DHBV infectable tissue culture systems do exist. There are, however, significant differences between the duck and human hepatitis viruses. For example, and perhaps of greatest relevance, human HBV envelope polypeptides, the likely mediators of entry, are N-glycosylated, whereas DHBV envelope polyepotides are not. Thus, the degree to which information from DHBV applies to human HBV attachment and entry may be limited.
The HBV surface protein antigens (HBsAg) are comprised of three carboxyl-co-terminal HBs proteins termed large (LHBs), middle (MHBs) and small (SHBs, also called major) protein [3, 4]. LHBs and MHBs also share the highly hydrophobic, repetitive, membrane-spanning S domain. In addition, MHBs has a 55 amino acid region called preS2, LHBs has an additional 109-120 amino acid long region called preS1 (dependent on the viral subtype) in their N-terminus (figure 1 A) [4]. Although HBV surface proteins must certainly mediate early steps in the virus life cycle, the precise role for each glycoprotein in the entry and egress of the virus is controversial. We will thus discuss this now.
LHBs is essential for attachment has been generally accepted. The studies of hepadnavirus in cell culture, especially with explanted human or duck primary liver cells, strongly suggest that LHBs is directly involved in the viral attachment [18, 19, 20]. The putative attachment site of HBV located in the preS1 was first reported by Neurath and his colleagues using anti-preS1 antibody [21, 22]. They found that the antibody against peptide corresponding to the HBV preS1 domain was virus-neutralizing and protective [21]. In 1989, Pontisso and his colleagues, using the membranes of surgically obtained human liver as a target, further confirmed the role of LHBs in the HBV attachment [23]. Recently, the attachment site of LHBs was functionally narrowed down to the amino acids 21–47 of preS1 by employing synthetic peptides. The results suggest that this binding site was not only required but also sufficient to attach specifically HepG2 cells (Figure 1 B) [24, 25]. Moreover, the antibody with this site has neutralized the HBV infection in Chimpanzee [19, 21]. It is interesting that, by mutagenesis studies and single cell attachment analysis, Paran and his colleagues found that the QLDPAF sequence within this preS1 region was crucial for cell attachment [25]. Further evidence to support this hypothesis is that this sequence is also found in the other virus and bacterial functioning as an adhesion or attachment determinant [25]. This suggests that the QLDPAF sequence may have a more general role in viral infection.
There are differing reports about the role of MHBs in the generation of HBV in the transfected cells and in mediating HBV infection [8, 26]. MHBs seem to be absent in certain HBV variants from chronically infected patients [27, 28], suggesting a non essential function in the viral infection. On the other hand, misfolded MHBs seems to be able to interfere with the assembly of HBV virions in the persistently producing virus cell line HepG2.215 [29, 30]. Anti-preS2 antibody (that recognizes MHBs) can block HBV infection of primary human liver cell in vitro [31, 32] and chimpanzee liver cells in vivo [33] suggesting that MHBs are probably also involved in viral entry. HBV preS2 can specifically bind the poly-human albumin in vitro [34, 32, 35] and the mono human albumin in vivo [36, 37], more investigation is needed to determine whether this binding capability is involved in the viral attachment.
SHBs, the most abundant viral glycoprotein, certainly has a role in virus secretion [24]. Its role in virus uptake is supported by the studies of Leenders and his colleagues [38]. Using recombinant HBs protein, they have performed binding studies with adult human hepatocytes, rat hepatocytes, human fibroblasts, human peripheral blood mononuclear cells and plasma membranes derived from these cell types. The results suggest that SHBs was able to bind specifically to human hepatocytes, human fibroblasts and human blood mononuclear cells but not the rat hepatocytes. This binding could be inhibited by recombinant HBs but not by the recombinant LHBs. The binding of SHBs with human hepatocytes was further supported by the observation from Bruin et al [39]. They have found that the particles only composed by SHBs can bind the human hepatocyte plasma membrane and this binding resulted in the internalization of the gold labeled sub viral particles [25].
However, despite the literature about HBV surface proteins, determination of which of the proteins is centrally responsible for HBV attachment and entry is elusive. Recently, synthetic beads conjugated to viral proteins were used to quantitatively measure virus–cell attachment, at the level of single cell resolution, by light microscopy [25]. In addition to attachment, entry of the HBV–protein conjugated beads into the cells can be easily detected by scanning and transmission electron microscopy. It was found that the beads conjugated with the recombinant preS1 protein containing the prominent QLDPAF motif, but lacking the small HBsAg regions, show efficiently attach to cells. Interestingly, beads conjugated with particles containing the whole repertoire of the surface proteins were twice as active in attachment, compared to preS1 alone. Thus, it appears that it is possible HBV uptake involves multiple attachment sties [17].
3. The cell receptor for HBV attachment
It is also unclear as to the identity of the cellular macromolecules that are responsible for virus attachment. Since HBV is highly infectious, in vivo, the relative refractoriness of liver cells, grown in culture, has been something of a puzzle. The Studies duck hepatitis B virus (DHBV) infection with primary duck hepatocytes have been particularly revealing. In 1994, then in 1995, Kuroki et al. reported that a glycoprotein in duck hepatocytes with the molecular weight 180 Kilo Dalton, called gp180, can be co-precipitated with DHBV [40, 41]. Moreover, the interaction of gp180 and DHBV can be blocked by monoclonal anti duck large protein antibodies. Subsequent cDNA cloning revealed the binding protein to be a carboxypeptidase H-like molecule, now classified as duck carboxypeptidase D (DCPD). Li and Tong have confirmed those [42, 43] and further demonstrated the carboxypeptidase D, a protease existing in the avian hepatocyte, exiting in the mammalian hepatocytes either. The protease function of carboxypeptidase D during the DHBV infection is unclear. The DHBV attachment receptor is a protease implies that hepadnavirus infection probably requires proteolysis. This will be discussed later.
The study to identify the receptors for human HBV LHBs has been less successful. Despite the fact that the specific preS1 domain for attachment of the LHBs to human liver cells and/or cell lines has been repeatedly verified, the cellular protein that is responsible for this binding has not been found yet. The limited availability of primary human tissue almost certainly hampers progress. Research has thus relied heavily upon human hepatoma cell lines [44]. Using a synthesized peptide with the sequence of HBV putative attachment site in preS1, Falco et al. have isolated a membrane protein called Hep-BP from HepG2 cells, which can specifically bind HBV preS1 peptide. Furthermore, they found that after the transfection of corresponding cDNA of Hep-BP into HepG2 cells, the virus binding capacity increased by 2 orders of magnitude compared with untransfected cells. Chinese hamster ovary cells, which normally do not bind to HBV, acquired susceptibility to HBV binding after transfection [45]. The sequence analysis suggests this protein is highly homologous with the squamous cell carcinoma (SCCA1), a serine protease inhibitor of ovalbumin family that has been used as a tumor marker for diagnosis of SCC [46]. However, the lack of the tissue specificity and low level expression in the liver cells reduce the possibility of it as a relevant receptor of HBV infection. It is worthy that Hep-BP is a serine protease inhibitor and is over-expressed in the cancer cells [47]. The over-expression of another serine protease inhibitor kazal type (SPIK) in the HepG2 and Huh 7 cells was also observed by us. The possible role of this protease inhibitor in the HBV infection will be discussed later.
Using human hepatoma cell line or the membrane of human liver cells, the protein that bind HBV MHBs or SHBs has been reported by several groups. In 1989, Potisso et al. reported that human liver plasma membranes contain receptors for MHBs via associated with polymerized human serum albumin [32]. Subsequently, Buine et al identified human liver endonexin II (EII) as a specific SHBs binding protein [48]. A sialoglycoprotein was also reported as a receptor for SHBs binding in the uptake of HBV [49, 50]. However, it is noted that no direct evidence to show that the interaction of HBV with those receptors could result in the real infection. Therefore, the full understand of the HBV attachment and it receptors perhaps could only be achieved after a susceptible cell line has been established.
4. The HBV fusion and proteolysis
The attachment of HBV surface protein to the host cell is essential for HBV entry. However, attachment alone is not sufficient for viral infection. The fusion of viral protein and cell membrane allows the release of the viral genome into cytosol. Attachment and fusion are two distinct events. Attachment usually is receptor mediated, and therefore, is highly cell specific. In contrast, viral fusion is dependent on a specific domain within the viral surface protein, known as the "fusion motif,” which is normally composed of a series of hydrophobic amino acids [51, 52, 53, 54] and is independent of specific cell receptors and thus relatively less cell specific. Attachment possibly occurs at a low temperature of 4� C. However, fusion requires a temperature of at least 20� C.
In 1993, we reported that HBV has a conservative region in the N-terminus of its S domain, which encloses a hydrophobic sequence with 13 amino acids [51]. This sequence has all the characteristics of fusion motif of other human viruses such as paramyxoviruses and retroviruses including of HIV, RSV and influenza virus [5]. More important, this region includes a core hydrophobic sequence: FLG-LL-AG, which is supposed to trigger the viral–cell fusion [55, 56]. Notably, this sequence is conserved in all the hepadnavirus [5, 6]. The fusogenesis of this sequence was confirmed by Rodriguez-Crespo et al. Using the synthesized peptides containing this region of HBV and woodchuck hepatitis virus (WHV), they have demonstrated that these peptides can induce the fusion of bio-membranes, liposome leakage and haemolytic activity in vitro [57, 58]. We also have found that the peptides containing this sequence can trigger the syncytia of HepG2 cell from without (unpublished evidence). It is noteworthy that pre-digestion of HBV with a bacterial protease V8 allowed the HBV and WHV entry into the unsusceptible cell line such as HepG2 to initiate the infection [5, 6]. The reason that V8 protease treatment induces viral infectivity is not clear. However, V8 protease cleaves at a proteolytic sensitive region called “PEST region” that is just adjacent to the putative fusogenic domain of HBV suggests that the viral infection probably is the consequence of the exposure of HBV fusion domain (Figure 1 B) [5,6]. The proteolysis to make the viral fusion domain that does not locate at the N-terminus accessible for the infection was seen in the other virus [14, 59, 60].
The protease induced HBV infection can be enhanced by low pH. Our unpublished data showed that the binding of V8 digested HBV to the HepG2 cells was pH dependent. At a mild acid condition, for example, pH5.5, the binding ability of those HBV to Hep G2 cells was increased 60% than that at neutral condition. Considering of the possible exposure of HBV fusion domain by the digestion of V8 protease, this result implies that the low pH might help HBV fusion. Low pH helps viral fusion was observed in other human virus such as Semliki forest virus and Influenza virus in the endocytosis way [14, 13, 11, 61]. It is not clear whether the uptake of HBV requires the endocytosis. However, it was demonstrated that exposing the virus to the low pH triggers translocation of the internally sequence of PreS1 domain onto the viral surface [62, 63]. This supports the hypothesis that the low pH in the endosome may induce the conformation change of HBV surface proteins, furthermore, to release its nucleocapsid after viral-cell fusion.
The results on the role of pH in the uptake of DHBV are inconsistent [64, 65]. Using lysosometropic agents, for example, ammonium chloride and chloroquine, which can raise the pH in the endosome, Offensperger et al. found that the uptake of DHBV has been blocked [64]. In contrast, Rigg et al. reported that the treatment of ammonium chloride did not affect the uptake of DHBV [65]. In spite both groups used the duck primary liver cell as an experimental infection system, however, the condition of the cell growth seems somehow different. Therefore, the contradictory observation might be the result of the different approaches used. Recently, Breiner and Schaller have demonstrated that the uptake of DHBV by primary duck hepatocyte (PDH) requires endocytosis [66]. The deficient mutant preventing the transport of DHBV to endosome has abolished the infection. However, the endocysis is a feature usually associated with low pH-triggered fusion mechanism. Altogether, while a strict low pH dependency may not apply to DHBV to the same extent as for certain other viruses, low pH may still significantly facilitate entry of the virus [17].
5. Internalization of HBV following attachment
Most data about hepadnavirus internalization and transport to the nucleus was generated from the studies of the uptake of DHBV by duck primary hepatocytes (PDH). However, the entry route for DHBV is still unclear. As we have mentioned before, there are two entryways that the viruses use for their entry. One is via direct fusion with the host cell membrane like HIV [67] and the other is via endocytic pathway like Semliki forest virus and Influenza virus [13, 14]. Recent data suggests that DHBV may use the endocytosis way for its entry [66]. This hypothesis was supported by the observation of the conformation change of the large surface protein of DHBV at low pH condition [62]. However, the more detail investigation was from the kinetic study of the uptake of DHBV by duck primary hepatocyte. In 1999, Qiao and his colleagues have performed a kinetic study of DHBV uptake. They observed that DHBV DNA appeared in the cytosol of PDH as early as one hour after post-incubation of virus with cells at 37 �C [68]. Nevertheless, DHBV DNA only can be detected in the nuclear fractions after at least 4 hours post-incubation. This suggests that the internalization happens immediately after the viral attachment. However, the intranuclear translocation of DHBV requires more time. The cccDNA and single-stranded DNA were not detected until 48 hours post inoculation (p.i.) [68]. Comparing with the in vivo infection, cccDNA can be detected in hepatocytes as fast as 6 hours after inoculation of ducks with the same DHBV strain [68], there was 40 hours delay during DHBV localization to the nucleus and formation of cccDNA in vitro system [68]. The reason for this delay is unknown.
Until now, there is limited data about HBV internalization and transport to the nucleus. However, the study of HBV infection to human primary hepatocytes shows that the cccDNA of HBV appeared in the nucleus after two days of the post inoculation, this is agreement with the observation from DHBV [69]. The result from the kinetic study on the internalization and transportation of HBV by the HepG2 cells is somehow different with the result from the primary hepatocytes. HepG2 is capable of supporting viral replication and producing progeny viruses. However, this cell-line is generally refractory with the infection of serum derived HBV [5, 15] in spite the post attachment internalization of HBV in HepG2 cells was observed by several groups [15, 49]. According to the observation from the Qiao's group, the internalization of HBV by HepG2 cells happened immediately after the viral attachment, and reached its peak within the cytosol two hours after post incubation at 37 �C. This was similar to the process in DHBV infection. However, the nuclear transport of HBV DNA was not completed even 16 hours following viral absorption [15]. The disability of transport of HBV DNA to nucleus implies that the release of HBV nucleocapsid in the HepG2 cells was blocked by some unknown facts after the viral internalization. Perhaps deficiencies in viral transport to the nucleus are part of the reason that HepG2 cells are refractory to HBV infection. Considering the proteolysis enhances in vitro HBV infection of HepG2 cells, it is possible that the block of HBV nucleocapsid transport is a consequence of the lack of necessary protease digestion.
It is still unclear whether the internalization of HBV requires a permeable domain with the sequence of PLSSIFSRIGDP at middle of preS2, which was reported by Oess and his colleagues recently (Figure 1B) [70]. It was shown that this domain mediated the internalization of a fused protein into Hela cells [70]. CD spectroscopy revealed the sequence of PLSSIFSRIGDP consists of a hydrophobic region that contains internal, charged, hydrophilic residues and had the ability to fold into an amphipathic alpha-helix, which has been identified conferring the unexpected property of cell permeability in other viruses [71]. Examples include the antennapedia homeo domain [53], HIV-tat transcription factor [72], Herpes simple virus vp22 [73] and lactoferrin [74]. It is interesting that this permeable domain is just adjacent with PEST-fusion region of HBV (Figure 1 B). The role of this permeable domain in HBV internalization needs further investigation.
6. Over-expression of Serine protease inhibitor Kazal in the HBV insusceptible hepatoma cell lines
If the refractoriness of HepG2 cells to HBV infection is related to the lack of requisite proteolysis, the conclusion might be that the HepG2 cells perhaps do not have these specific proteases or they do have these proteases, however, they are inactivated by an unknown agent. To find clues as to the nature of HepG2 refractoriness to HBV infection, we have compared the gene expression profile of HBV susceptible primary human liver and HepG2 cells, with an eye on expression of RNA specifying proteases and their inhibitors, using a gene array system. The results show that there were no detected differences in the protease profile. However, the expression of a serine protease inhibitor Kazal (SPIK) was more than a thousand fold higher in HepG2 cells than in human liver cells (unpublished data). At the same time, the expressions of the most other genes were found to be approximately equal amongst HepG2 cells and human liver cells (unpublished gene array data). This suggests a role for SPIK in the altered phenotype of HepG2 cells with respect to liver cells and possibly a role in their susceptibility differences to HBV infection. These finding that SIPK was over expressed in HepG2 cells, relative to primary liver tissue, was confirmed by reverse-transcription PCR (RT-PCR) and Northern blot analysis.
As shown in figure 2, two specific primer sets, PI1/ PI2 and PI3/PI4, were used to amplify SPIK mRNA by RT-PCR. PI1/PI2 directly amplifies the SPIK gene from the 11th bp to233th bp and PI3/PI4 directly amplifies the entire SPIK gene (Figure 2A). The RT-PCR band generated from primers PI3/PI4 was at the predicted size and slightly larger than that one generated from primers PI1/PI2 (324 base pairs versus 224 base pairs (Figure 2B). This suggests that the reactions of RT-PCR were specific. In addition, these bands were not generated from SPIK DNA (gene sequences), because the control that lacked the enzyme reverse transcriptase in the reaction mixture did not show any band (Figure 2B, lane 9). Compared to human liver cells, the expression of SPIK gene in the HepG2 cells was overwhelming. Using either of the primer sets, SPIK specific bands can be easily detected in the HepG2 cells, but hardly in the human liver cells (Figure 2B). This difference was not a result of the variation of loading, because the expression of the thimet oligopeptidase gene in the same condition did not show any difference (Figure 2C).
The over-expression of the SPIK gene in HepG2 cells has also been confirmed by Northern blot analysis. Ten micrograms RNA from HepG2 cells and human liver cells were resolved on a 1% non-denatured agarose gel, subsequently transferred to a nylon membrane, and then hybridized with a 32P labeled SPIK specific probe derived from the SPIK gene by PCR. In agreement with the results from RT-PCR, figure 3 shows that the SPIK mRNA can only be detected in the HepG2 cells. The signal in the human liver cells was invisible (Figure 3, Northern blot, lane 2 & lane 3). The undetectable signal from human liver cells was not as a result of the unequal loading. The equality of RNAs were applied on to the gel as evidenced by the equivalent ethidium bromide (EB) staining of ribosomal RNA in both samples (Figure 3, EB stain lane 2 and lane 3. See 18S and 26S ribosome RNA). Similar increase of the SPIK expression was observed in yet another hepatoma cell line Huh7, which is also refractory to HBV infection similarly to HepG2 (Figure 3. Northern blot, lane 1). Taken together, these results demonstrate that SPIK gene is over-expressed in cells that are not susceptible to HBV infection, such as HepG2 and Huh7 cells. In contrast, the expression of SPIK maintains at undetectable level in the HBV infectible cells such as the normal human liver cells. Although, this needs to be further investigated, it is also worth noting that if over expression of SIPK is common to hepatoblastoma and other cancer cells, it could be a useful biomarker for early detection of cancer (Lu, in progress).
7. Systems used for the study of HBV infection
As we have mentioned before, the major limitation to study HBV entry is the lack of an in vitro infection system, which can support the entire life cycle of HBV. Human primary hepatocyte cultures derived from liver explants have been shown to be susceptible to HBV infection, but only for a limited amount of time following explantation [75, 76]. Besides of the human primary hepatocytes, there are several systems derived from Hepatoma cells so far were used for the study of HBV infection in vitro [68, 77, 78, 16, 79]. Unfortunately, no one of them is perfect. This includes the manipulation of the cultured HepG2 cells with dimethyl sulfoxide (DMSO) or polyethyleneglycol (PEG), which has been reported to enhance HBV infection in vitro [68, 77]. Currently, a cell line susceptible to HBV infection was reported by Grippon et al. [16]. However, the high dose of DMSO and 2-4% PEG were used in this system to maintain susceptibility greatly limits their application for study the natural infection of HBV. Therefore, although cultured cells can be shown to support HBV replication, there is yet no readily reproducible tissue culture system for HBV infectivity.
The finding of over expression of SPIK in the HepG2 cells and Huh 7 cells may suggest a strategy to establish a stable cell line that is susceptible for HBV infection by silencing those over expressed proteases inhibitor. Admittedly, the cause and effect relationship between over expression of SIPK and refractoriness to HBV is a long shot. Never the less, it could be tested by straightforward techniques that were in use in the lab for other purposes. Antisense oligonucleotides have been used previously to silence specific genes [80]. In one experiment, HepG2 cells were pre-treated with 10 nM anti-sense or sense oligo corresponding to the translation initiation region of SPIK gene. After 2 days, cells were then infected with HBsAg positive patient's serum (107 viruses /ml). The progeny HBV both in the medium and in the cytosol were examined. By 3 days after the inoculation, the amount of HBsAg in the culture medium fell below the level of detection. However, the level of HBsAg in the culture medium from HepG2 cells pre-treated with anti-sense of SPIK significantly increased on the 4th day and 5th day post infection (p.i.), then decreased but maintained in a detectable level until harvest (7 days post infection, figure 4). On the other hand, HBsAg in the culture medium from cells that were treated with sense oligo of SPIK were at background levels during entire period (figure 4). A similar result was found in the untreated HepG2 cells (data not shown).
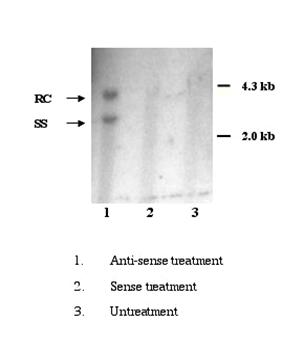
HepG2 cells were susceptible to HBV infection after treatment with SPIK specific anti-sense oligo was confirmed by the results from Southern blot. At 8th day post infection, the replicative form of HBV in cytoplasm was detected feebly in infected cells that were pre-treated with anti-sense of SPIK (Figure 5. lane 1). However, no HBV DNA was found in cells treated with SPIK sense oligo or when untreated (Figure 5. lane 2, 3). The examining cccDNA in the nuclear fraction of infected cells was unsuccessful. It is likely that the levels were far below the detection limit.
In spite the signals from infected cells were not very intense (this may be due to the less efficiently transport of anti-sense oligo to the cells), but were reproducibly present. However, the fact that HepG2 cells appear to be successfully infected by HBV following marginal down regulation of SPIK gene expression supports our hypothesis that the suppression of the over-expression of SPIK gene might reinstate the susceptibility of HepG2 cell to HBV infection by the restoration of the activity of indispensable protease. Currently, a new technology called RNA interference (RNAi) or short interfering RNA (siRNA) was developed to silence target gene expression [81, 82, 83]. Using this method a stable cell line, in which the SPIK gene is silenced, could be established by using of the specific siRNA, and then be selected by co-expressing a selectable gene such as the neo gene through screening with G418 [84]. After screening with G418, all surviving cells would be expected to generate the specific siRNA silencing the targeted SPIK expression. The early results are, never the less, provocative, and beg further investigation. An effort, for example, to establish a susceptible cell line to HBV infection by silencing SIPK is now under way.
Acknowledgements
This work is supported by Hepatitis B Foundation of America; an Appropriation from the Commonwealth of Pennsylvania USA, Nucleonic Inc. (PA USA). Drs. Satishchandran C and Cathy Pachuk (Nucleonics Inc, PA, USA), Baohua Gao and Tienlun Zhou (Thomas Jefferson university, USA) are thankful for their comments.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Seeger C, Mason WS. Hepatitis B virus biology. Microbiology & Molecular Biology Reviews. 2000 ;64 (1) :51 -68
2. Mason WS, Seeger C. Hepadenavirus-molecular biology and pathogenesis. Curr. Top. Microbiol. Immunol. 1991 :- 168
3. Stibbe W, Gerlich WH. Structural relationships between minor and major proteins of hepatitis B surface antigen. J. Virol. 1983 ;46 :626 -629
4. Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. Large surface proteins of hepatitis B virus containing the pre-S sequence. J. Virol. 1984 ;52 :396 - 402
5. Lu X, Block T, Gerlich WH. Protease-induced infectivity of hepatitis B virus for a human hepatoma cell line. J. Virol. 1996 ;70 :2277 -2285
6. Lu X, Hazboun T, Block T. Limited proteolysis induces woodchuck hepatitis virus infectivity for human HepG2 cells. Virus Research. 2001 ;73 (1) :27 -40
7. Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986 ;47 :451 -460
8. Ueda K, Tsurimoto T, Matsubara K. Three envelope proteins of Hepatitis B virus: Large S, Middle S, and major SHBs needed for the formation of Dane particles. J. Virol. 1991 ;65 (7) :3521 -3529
9. Birnbaum F, Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. Journal of virology. 1990 ;64 :3319 -3330
10. Huovila AJ, Eder AM, Fuller SD. Hepatitis B surface antigen assemblers in a post-ER, pre-Golgi compartment. Journal of Cell biology. 1992 ;118 :1305 -1320
11. Lonberg-Holm K, Philipson L. Virus receptor: part 2- animal viruses. In: (ed.) Lonberg-Holm K, Philipson L. Receptors and recognition series B Vol 8. London: Chapman and Hall. 1981 :85-211
12. Debra M, Eckert A, Peter S. Mechanism of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001 ;70 :777 -810
13. Lobigs M, Garoff H. Fusion function of Semliki Forest virus spike is activated by proteolytic cleavage of the envelope glycoprotein precursor P62. J. Virol. 1990 ;64 :1233 -1240
14. Formanowski F, Warton SA, Calder CJ, Hofbaner C, Meier-Ewert H. Fusion characteristic of influenza C virus. J. Gen. Virol. 1990 ;71 :1181 -1188
15. Qiao M, Macnaughton TB, Gowans EJ. Adsorption and penetration of hepatitis B virus in a nonpermissive cell line. Virology. 1994 ;201 :356 -363
16. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Infection of a human hepatoma cell line by hepatitis B virus. PNAS USA. 2002 ;99 (24) :15655 -60
17. Cooper A, Paran N, Shaul Y. The earliest steps in hepatitis B virus infection. EMBO J. 2003 ;13 :2273 -2279
18. Glebe D, Aliakbari M, Krass P, Knoop EV, Valerius KP, Gerlich WH. Pre-s1 antigen-dependent infection of Tupaia hepatocyte cultures with human hepatitis B virus. J Virol. 2003 ;77 (17) :9511 -9521
19. Hong HJ, Ryu CJ, Hur H, Kim S, Oh HK, Oh MS, Park SY. In vivo neutralization of hepatitis B virus infection by an anti-preS1 humanized antibody in chimpanzees. Virology. 2004 ;318 (1) :134 -141
20. Urban S, Gripon P. Inhibition of duck hepatitis B virus infection by a myristoylated pre-S peptide of the large viral surface protein. J Virol. 2002 ;76 (4) :1986 -1990
21. Neurath AR, Seto B, Strick N. Antibodies to synthetic peptides from the PreS I region of the hepatitis B virus (HBV) envelope (env) protein are virus neutralizing and protective. Vaccine. 1989 ;7 :234 -236
22. Neurath AR, Kent SB, Strick N, Parker K. Identification and chemical synthesis of a host cell receptor binding site on hepatitis B virus. Cell. 1986 ;46 (3) :429 -36
23. Pontisso P, Ruvoletto MG, Gerlich WH, Heermann KH, Bardini R, Alberti A. Identification of an attachment site for human liver plasma membranes on hepatitis B virus particles. Virology. 1989 ;173 (2) :522 -530
24. Bruss V, Ganem D. The role of envelope proteins in hepatitis B virus assembly. PNAS USA. 1991 ;88 :1059 -1063
25. Paran N, Geiger B, Shaul Y. HBV infection of cell culture: evidence for multivalent and cooperative attachment. EMBO J. 2001 ;20 :4443 -4453
26. Bruss V, Ganem D. Mutational analysis of hepatitis B surface antigen particle assembly and secretion. Journal of Virology. 1991 ;65 :3813 -3820
27. Fernholz D, Galle PR, Stemler M, Brunetto M, Bonino F, Will H. Infectious hepatitis B virus variant defective in pre-S2 protein expression in a chronic carrier. Virology. 1993 ;194 (1) :137 -148
28. Pollicino T, Zanetti AR, Cacciola I, Petit AM, Smedile A, Campo S. Pre-S2 defective Hepatitis B Virus infection in patients with fulminant hepatitis. Hepatology. 1997 ;26 :495 -499
29. Lu X, Lu Y, Geschwindt R, Dwek RA, Block TM. Hepatitis B virus MHBs antigen is selectively sensitive to glucosidase-mediated processing in the endoplasmic reticulum. DNA Cell Biol. 2001 ;20 (10) :647 -656
30. Lu X, Mehta A, Dwek R, Butters T, Block T. Evidence that N-linked glycosylation is necessary for hepatitis B virus secretion. Virology. 1995 ;213 :550 -665
31. Ryu CJ, Gripon P, Park H, Park S, Kim K, Guguen-Guillouzo C, Yoo O, Hong H. In Vitro Neutralization of Hepatitis B Virus by Monoclonal Antibodies Against the Viral Surface Antigen. J. Med. Virol. 1997 ;52 :226 -233
32. Pontisso P, Petit MA, Bankowski MJ, Peeples ME. Human liver plasma membranes contain receptors for the hepatitis B virus pre-S1 region and, via polymerized human serum albumin, for the pre-S2 region. J Virol. 1989 ;63 (5) :1981 -8
33. Neurath AR, Kent SB, Parker K, Prince AM, Strick N, Brotman B, Sproul P. Antibodies to a synthetic peptide from the preS 120-145 region of the hepatitis B virus envelope are virus neutralizing. Vaccine. 1986 ;4 (1) :35 -7
34. Machida A, Kishimoto S, Ohnuma H, Miyamoto H, Baba K, Oda K, Nakamura T, Miyakawa Y, Mayumi M. A hepatitis B surface antigen polypeptide (P31) with the receptor for polymerized human as well as chimpanzee albumins. Gastroenterology. 1983 ;185 (2) :268 -74
35. Pontisso P, Falcieri E, Schiavon E, Alberti A, Realdi G. Polyalbumin receptors on hepatitis B virus and on 22 nm hepatitis B surface antigen (HBsAg)2 particles. J Med Virol. 1984 ;13 (4) :355 -360
36. Krone B, Lenz A, Heermann KH, Seifer M, Lu XY, Gerlich WH. Interaction between hepatitis B surface proteins and monomeric human serum albumin. Hepatology. 1990 ;11 (6) :1050 -1056
37. Lu X, Yao GB, Tien YF. The Interaction between Native Serum Albumin and Hepatitis B Virus. Arch. of Virol. 1988 ;98 :163 -170
38. Leenders WP, Glansbeek HL, de Bruin WC, Yap SH. Binding of the major and large HBsAg to human hepatocytes and liver plasma membranes: putative external and internal receptors for infection and secretion of hepatitis B virus. Hepatology. 1990 ;12 (1) :141 -147
39. Bruin WC, Hertogs K, Leenders WP, Depla E, Yap SH. Hepatitis B virus: specific binding and internalization of small HBsAg by human hepatocytes. J Gen Virol. 1995 ;76 (4) :1047 -50
40. Kuroki K, Cheung P, Marion L, Ganem D. A cell surface protein that binds avian hepatitis B virus particles. J. Virol. 1994 ;68 :2091 -2096
41. Kuroki K, Eng F, Ishikawa T, Turck C, Harada F, Ganem D. gp180, a host cell glycoprotein that binds duck hepatitis B virus particles, is encoded by a member of the carboxypeptidase gene family. J. Biol. Chem. 1995 ;270 :15022 -15028
42. Li J, Tong S, Wands JR. Characterization of a 120-kilodalton pre-S binding protein as a candidate duck hepatitis B virus receptor. J. Virol. 1996 ;70 :6029 -6038
43. Tong S, Li J, Wands JR. Carboxypeptidase D is an avian hepatitis B virus receptor. J Virol. 1999 ;73 (10) :8696 -8702
44. Petit MA, Capel F, Dubanchet S, Mabit H. PreS1-specific binding proteins as potential receptors for hepatitis B virus in human hepatocytes. Virology. 1992 ;187 (1) :211 -222
45. Falco S, Ruvoletto MG, Verdoliva A, Ruvo M, Raucci A, Marino M, Senatore S, Cassani G, Alberti A, Pontisso P, Fassina G. Cloning and expression of a novel hepatitis B virus-binding protein from HepG2 cells. J Biol Chem. 2001 ;276 (39) :36613 -36623
46. Kato H. Clinical significance of SCC antigen assay as a tumor marker. Nippon Rinsho Japanese Journal of Clinical Medicine. 1997 ;48 (Suppl) :1057 -1059
47. Pontisso P, Calabrese F, Benvegnu L, Lise M, Belluco C, Ruvoletto MG, De Falco S, Marino M, Valente M, Nitti D, Gatta A, Fassina G. Overexpression of squamous cell carcinoma antigen variants in hepatocellular carcinoma. Br J Cancer. 2004 ;90 (4) :833 -837
48. Bruin W, Leenders W, Kos T, Hertogs K, Depla E, Yap SH. Hepatitis delta virus attaches to human hepatocytes via human liver endonexin II, a specific HBsAg binding protein. J Viral Hepat. 1994 ;1 (1) :33 -38
49. Treichel U, Meyer Z, Buschenfelde KH, Dienes HP, Gerken G. Receptor-mediated entry of hepatitis B virus particles into liver cells. Arch Virol. 1997 ;142 (3) :493 -498
50. Peeples ME, Komai K, Radek R, Bankowski MJ. A cultured cell receptor for the small S protein of hepatitis B virus. Virology. 1987 ;160 (1) :135 -142
51. Gerlich WH, Lu X, Heermann KH. Studies on the attachment and penetration of hepatitis B virus. J. Hepatol. 1993 ;17 (Suppl. 3) :S10 -S14
52. Hughson FM. Structural characterization of viral fusion proteins. Current Biology. 1995 ;5 (3) :265 -274
53. Kyte J, Doolittle RFA. Simple method for displaying the hydrophobic character of a protein. J Mol. Biol. 1982 ;157 :105 -132
54. Rose OD, Roy S. Hydrophobic basis of packing in globular proteins. PNAS USA. 1980 ;77 (8) :4643 -4647
55. Jason B, Scheller H. A fusion of new ideas. Nature. 1997 ;387 :133 -137
56. White JM. Membrane fusion. Science. 1992 ;258 :917 -924
57. Rodriguez-Crespo I, Nunez E, Gomez-Gutierrrez J, Yelamos B, Albar JP, Peterson DL, Gaivilanes F. Phospholipid interactions of the putative fusion peptide of hepatitis B virus surface antigenS. J. Gen. Virol. 1995 ;76 :301 -305
58. Rodriguez-Crespo I, Nunez E, Yelamos B, Gomez-Gutierrez J, Albar JP, Peterson DL, Gavilanes F. Fusogenic Activity of Hepadnavirus Peptides Corresponding to Sequences Downstream of the Putative Cleavage Site. Virology. 1999 ;261 :133 -142
59. Nagai Y. Virus activation by host proteinases: A pivotal role in the spread of infection, tissue tropism and pathogenicity. Microbiology & Immunology. 1995 ;39 (1) :1 -9
60. Sturzenbecker L, Nibert M, Furlong D, Fields BN. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J. Virol. 1987 ;61 :2351 -2236
61. Gaudin Y, Ruigrok WH, Brunner J. Low-pH induced conformation change in viral fusion proteins: implications for the fusion mechanism. J. Gen. Virol. 1995 ;76 :1541 -1556
62. Guo JT, Pugh JC. Monoclonal antibodies to a 55-kilodalton protein present in duck liver inhibit infection of primary duck hepatocytes with duck hepatitis B virus. J. Virol. 1997 ;71 (6) :4829 -31
63. Bruss V, Lu X, Thomssen R, Gerlich WH. Posttranslational alterations in transmembrane topology. EMBO J. 1994 ;13 :2273 -2279
64. Offensperger WB, Offensperger S, Walter E, Blum HE, Gerok W. Inhibition of duck hepatitis B virus infection by lysosomotropic agents. Virology. 1991 ;183 (1) :415 -418
65. Rigg RJ, Schaller H. Duck hepatitis B virus infection of hepatocytes is not dependent on low pH. J Virol. 1992 ;66 (5) :2829 -2836
66. Breiner KM, Schaller H. Cellular receptor traffic is essential for productive duck hepatitis B virus infection. J Virol. 2000 ;74 (5) :2203 -2209
67. Dimitrov AS, Xiao X, Dimitrov DS, Blumenthal R. Early intermediates in HIV-1 envelope glycoprotein-mediated fusion triggered by CD4 and co-receptor complexes. Journal of Biological Chemistry. 2001 ;276 (32) :30335 -41
68. Qiao M, Scougall CA, Duszynski A, Burrell CJ. Kinetics of early molecular events in duck hepatitis B virus replication in primary duck hepatocytes. J Gen Virol. 1999 ;80 (8) :2127 -2135
69. Gripon P, Diot C, Theze N, Fourel I, Loreal O, Brechot C, Guguen-Guillouzo C. Hepatitis B virus infection of adult human hepatocytes cultured in the presence of dimethyl sulfoxide. J. Virol. 1988 ;62 :4136 - 4143
70. Oess S, Hildt E. Novel cell permeable motif derived from the PreS2-domain of hepatitis-B virus surface antigens. Gene Ther. 2000 ;7 :750 -758
71. Hildt E, Urban S, Hofschneider PH. Characterization of essential domains for the functionality of the MHBst transcriptional activator and identification of a minimal MHBst transactivator. Oncogene. 1995 ;11 :2055 -2066
72. Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. PNSA USA. 1994 ;91 :664 -668
73. Elliott G, O'Hare P. Intercellular trafficking and protein delivery by a herpes virus structural protein. Cell. 1997 ;88 :223 -233
74. He J, Furmanski P. Sequence specificity and transcriptional activation in the binding of lactoferrin to DNA. Nature. 1995 ;373 :721 -724
75. Ochiya T, Tsurimoto T, Ueda K, Okubo K, Shiozawa M, Matsubara K. An in vitro system for infection with hepatitis B virus that uses primary human fetal hepatocytes. PNAS USA. 1989 ;86 (6) :1875 -1879
76. Galle PR, Haglestein J, Kommerell B. In vitro experimental infection of primary human heptocytes with hepatitis B virus. Gastroenterology. 1994 ;106 :664 -673
77. Gripon P, Diot C, Guguen-Guillouzo C. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology. 1993 ;192 :534 -540
78. Walter E, Keist R, Niederost B, Pult I, Blum HE. Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology. 1996 ;24 (1) :1 -5
79. Favre D, Petit MA, Trepo C. Latent hepatitis B virus (HBV) infection and HBV DNA integration is associated with further transformation of hepatoma cells in vitro. ALTEX. 2003 ;20 (3) :131 -42
80. Ehlers M. et al. Antisense oligonucleotide-mediated gene knockdown during thymocyte development reveals role for Runx3 transcription factor in CD4 silencing during development of CD4-/CD8+ thymocytes. Journal of Immunology. 2003 ;171 (7) :3594 -604
81. Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D melanogaster germline. Current Biology. 2001 ;11 (13) :1017 -27
82. Brenda LB. Double-Stranded RNA as a Template for Gene Silencing. Cell. 2000 ;101 :235 -238
83. Fire A. RNA-triggered gene silencing. Trends Genet. 1999 ;15 (9) :358 -63
84. Rees S, Coote J, Stables J, Goodson S, Harris S, Lee MG. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques. 1996 ;20 (1) :102 -104
Figures
Figure 1. The structure of Hepatitis B virus surface proteins and its interesting regions for infection. A. The structure of HBsAg. B. The location of the interesting regions for HBV infection.
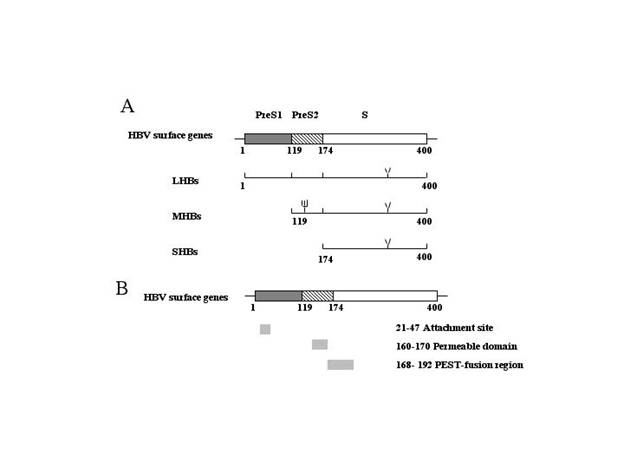
Figure 2. RT-PCR detects SPIK RNA. 1μg RNA from HepG2 cells and human liver cells were reversely transcripted and amplified by PCR using specific primer for SPIK. PCR bands were resolved in 1 % agarose gel and stain with ethidium Bromide. A: The names and location of primers. B: RT-PCR results. Lanes 1 to 4 were amplified with primers PI1/PI2; 5 to 8 were amplified with primers PI3/PI4. The samples from human liver cells were loaded in the column 1,2,5,6, (duplicated). The samples from HepG2 were loaded in the lane 3,4,7,8. Lane 9 was negative control without RTase. The specific SPIK bands are indicated by molecular weight. The lower bands in B are dimmer of primers. C: RT-PCR detection of TH gene of the human liver cells and HepG2 cells.
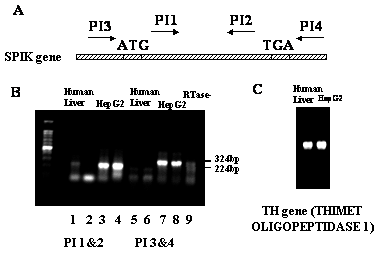
Figure 3. Northern blot detects SPIK. 10μg RNA from Huh7 (1), HepG2 (2) and human liver cells purchased from CloneTech (3) were resolved in the 1% non-denatured agarose gel, then, transferred to nylon membrane. The membrane was hybridized with 32p labeled SPIK specific probe. The image of ethidium bromide (EB) staining was taken from gel before transference. The image of northern blot was determined by phosphateImager.
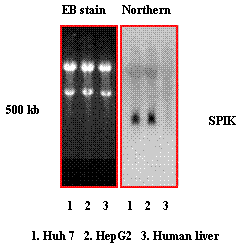
Figure 4. Secreted HBsAg in the HepG2 cells pre-treated with anti-sense of SPIK. HepG2 cells were pre-treated with 10nM/ml anti-sense or sense oligo of SPIK two days. Then, cells were infected with HBsAg positive patients' serum. The secreted HBsAg in the culture medium was detected by Auszyme kit (Abbott Lab. Abbott Park, IL).
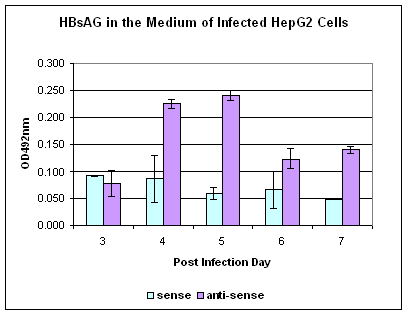
Figure 5. Southern blot detection of HBV DNA in the infected HepG2 cells. 106 HepG2 cells were pre-treated with anti-sense or sense oligo of SPIK or left untreated, then infected by patients' serum. Cells were harvested at 8th day p.i. After removal of nuclei, HBV DNA in cytoplasma was isolated, resolved in 1% agarose gel and detected by southern blot with HBV specific probe.
Author biography
Xuanyong Lu Ph. D is an Assistant Professor of Department of Biochemistry and Molecular Pharmacology, Thomas Jefferson University, Philadelphia, USA. His current researches investigate the entry of hepatitis B virus, particularly in the viral fusion and proteolysis. His researches also include the efforts to establish a cell line susceptible for HBV infection by silencing over expressed SPIK gene in the hepatoma cell lines.
Timothy M Block Ph. D is a Professor, director of Jefferson Center for Biomedical Research and Agricultural Medicine, Thomas Jefferson University, USA. His current researches include the role of glycan in the HBV infection, assembly and secretion, the anti-HBV drug exploration and development and the latent of Herpes simple virus.
![]() Corresponding address:
Corresponding address:
Dr. Xuanyong Lu, Department of Biochemistry and Molecular Pharmacology, Thomas Jefferson University, 700 East Butler Ave, Doylestown, PA, 18901. Telephone: 215-489-4906. Fax: 215-489-4920. E-mail: Xuanyong.luedu.
Received 2004-2-02
Accepted 2004-3-03
Published 2004-3-10